Case of the Week #650
(1) Femicare, Center of prenatal ultrasonographic diagnostics, Martin, Slovakia; (2) Centro Médico Recoletas, Valladolid, Spain
A 35-year-old secundigravida was referred to our unit at 20 weeks gestation for evaluation of suspected fetal skeletal abnormalities. Her first child was healthy, and her personal and family history were otherwise unremarkable.
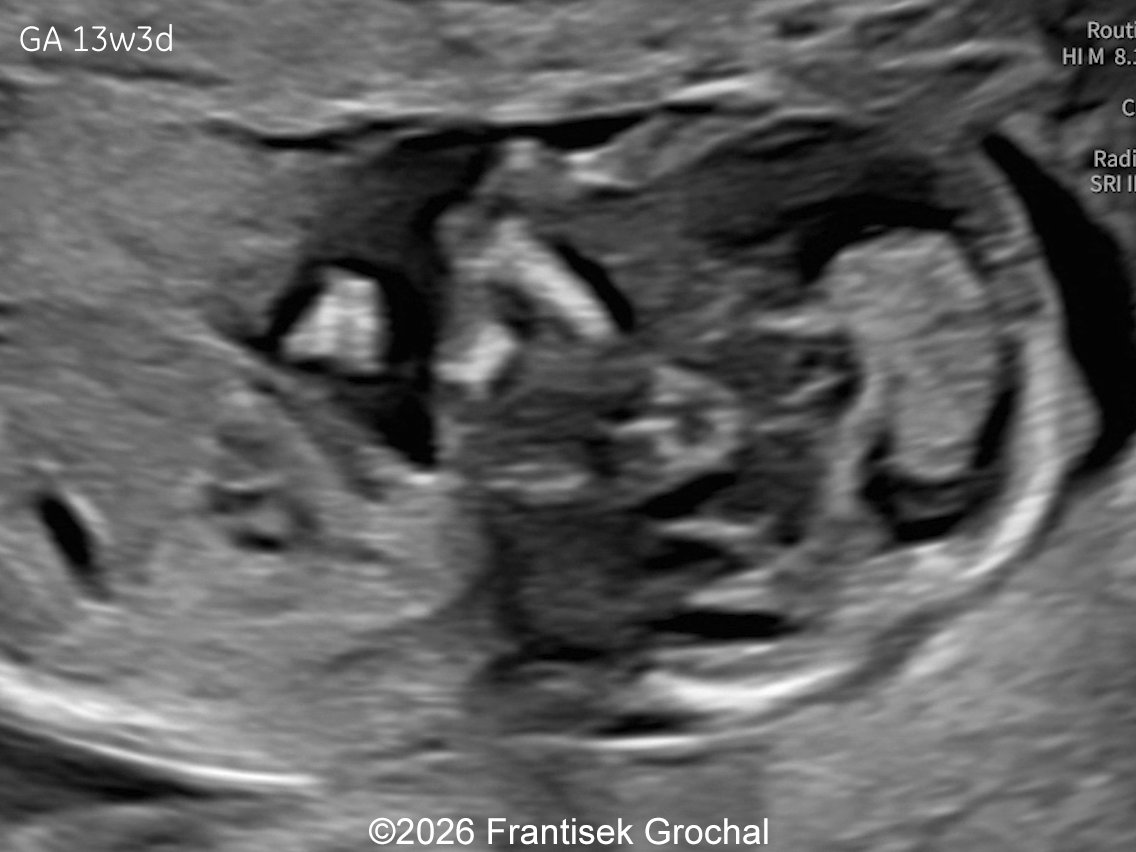
Two years later in a subsequent pregnancy, the patient presented for ultrasound evaluation. This examination was performed at 13 weeks of gestation and demonstrated the following findings:
View the Answer Hide the Answer
Answer
We present a case of Bruck syndrome.
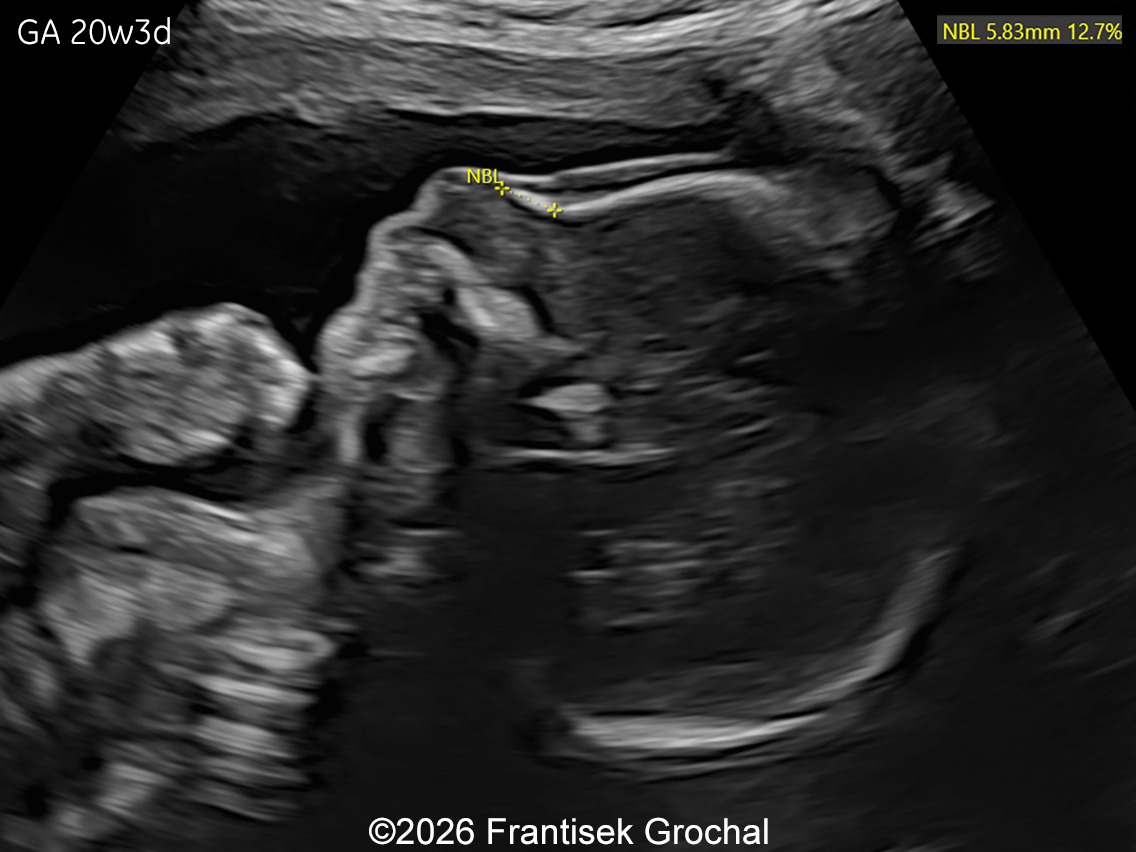
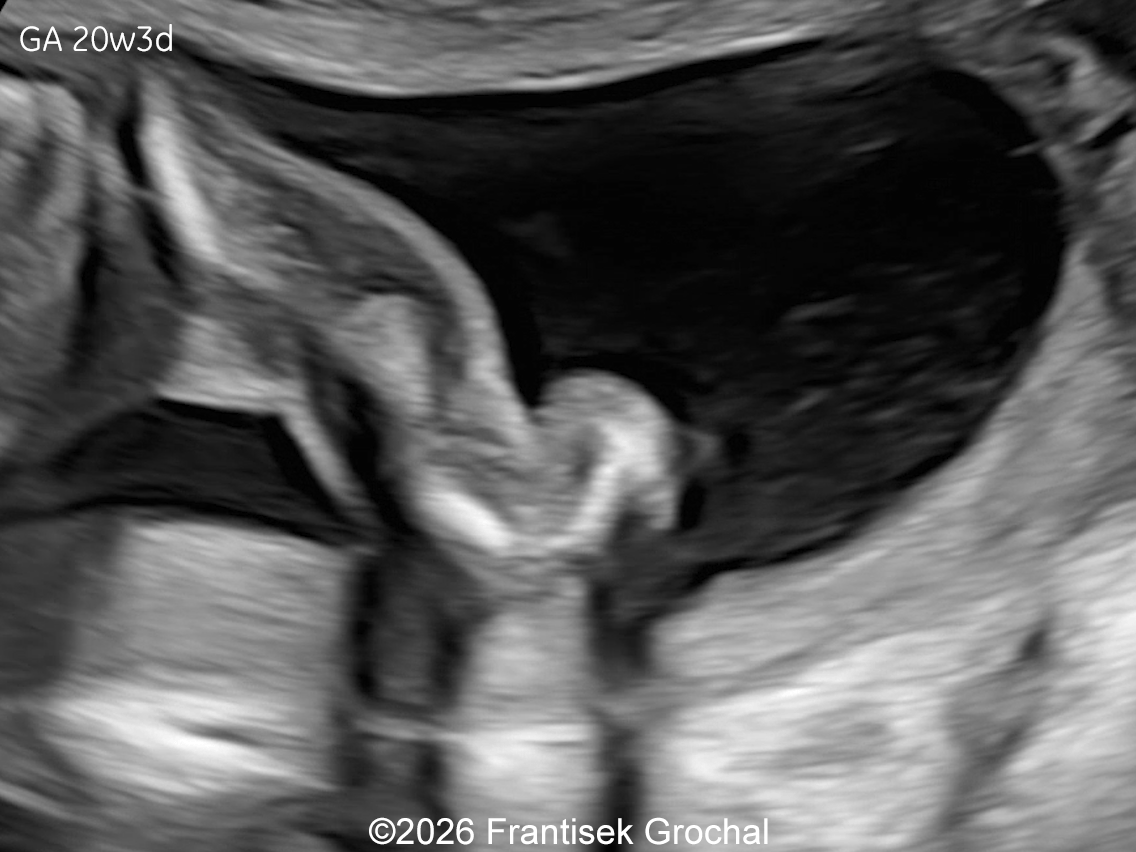
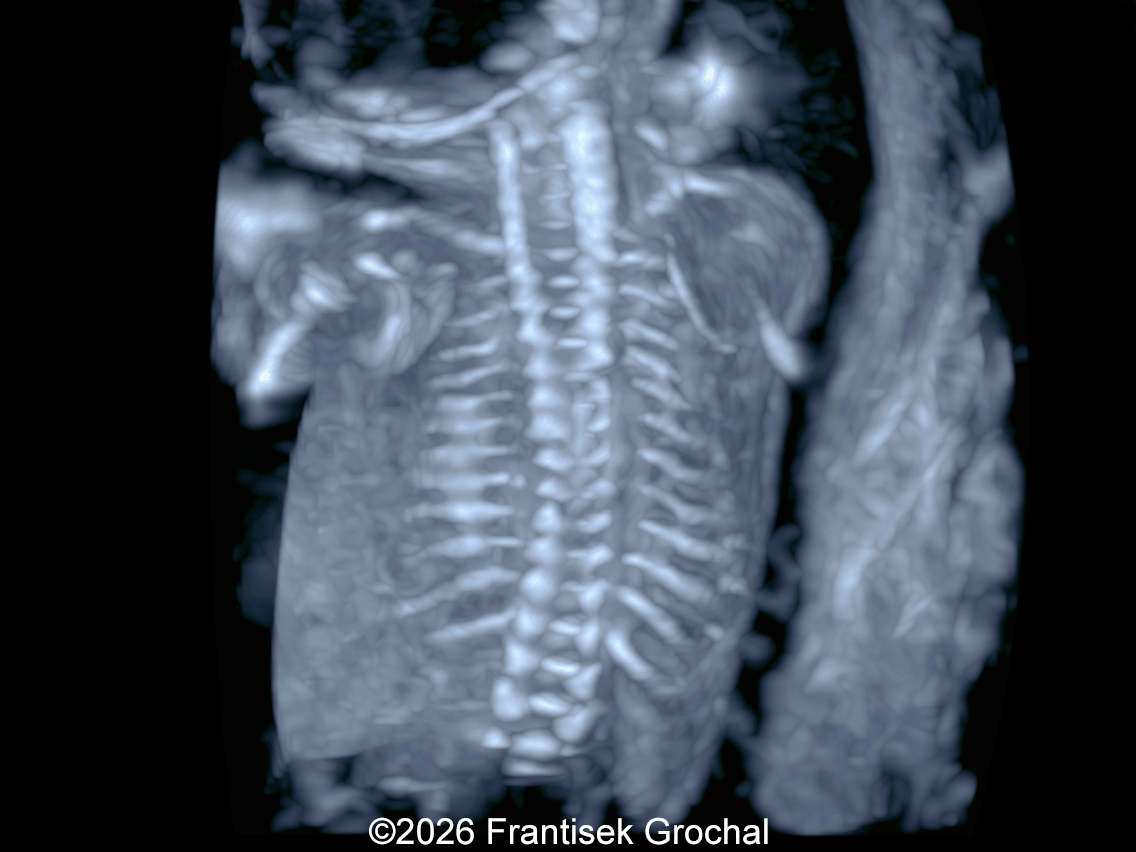
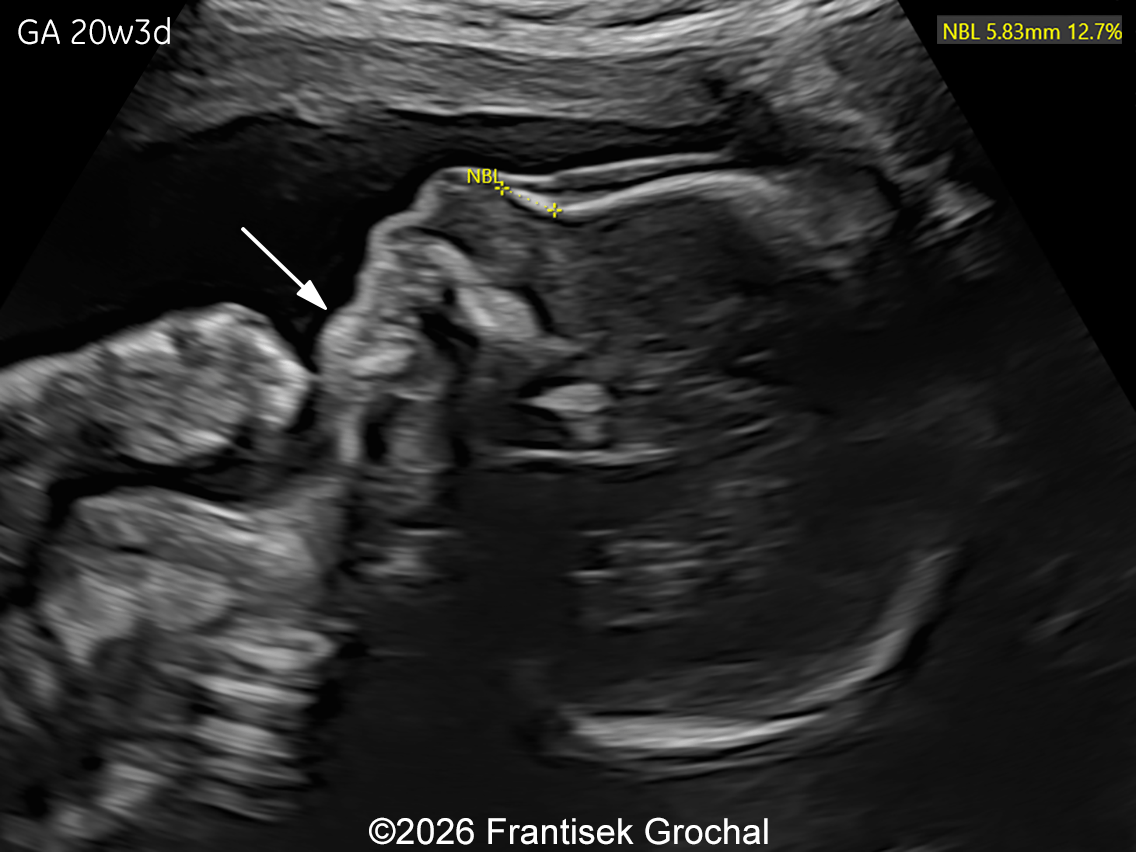
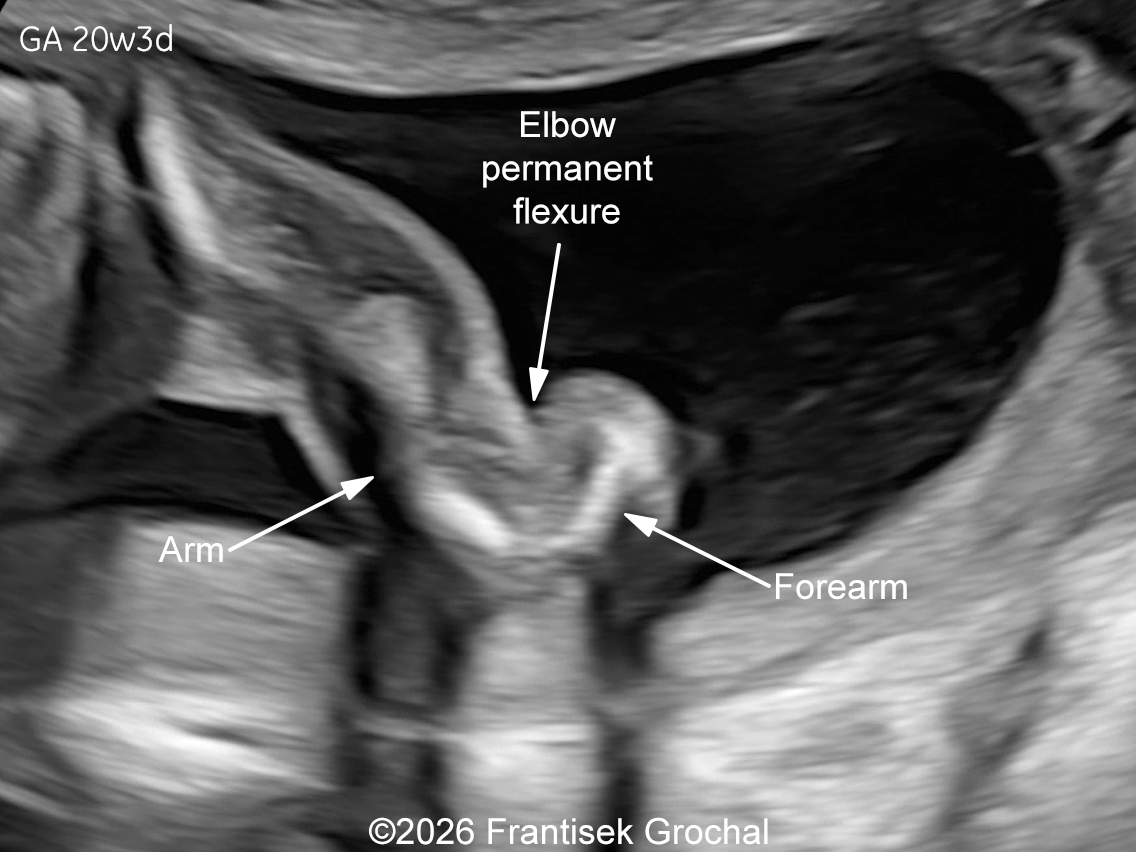
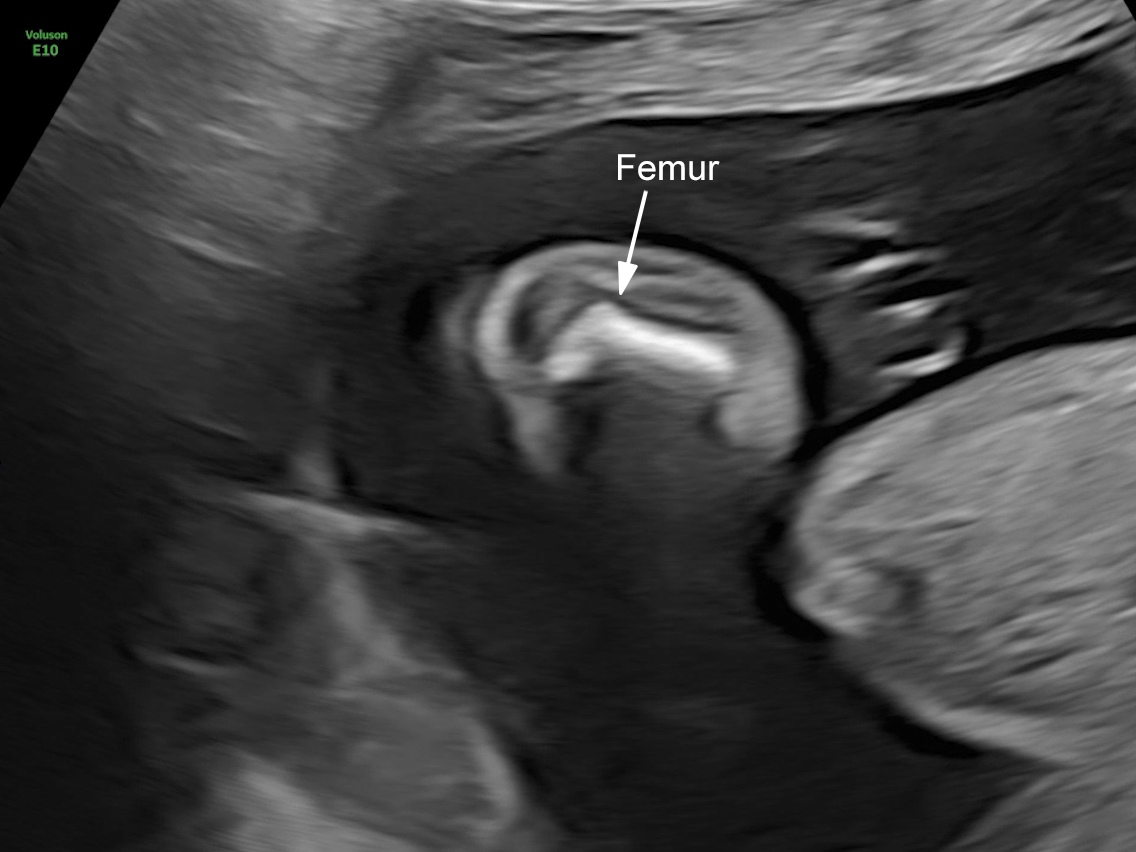
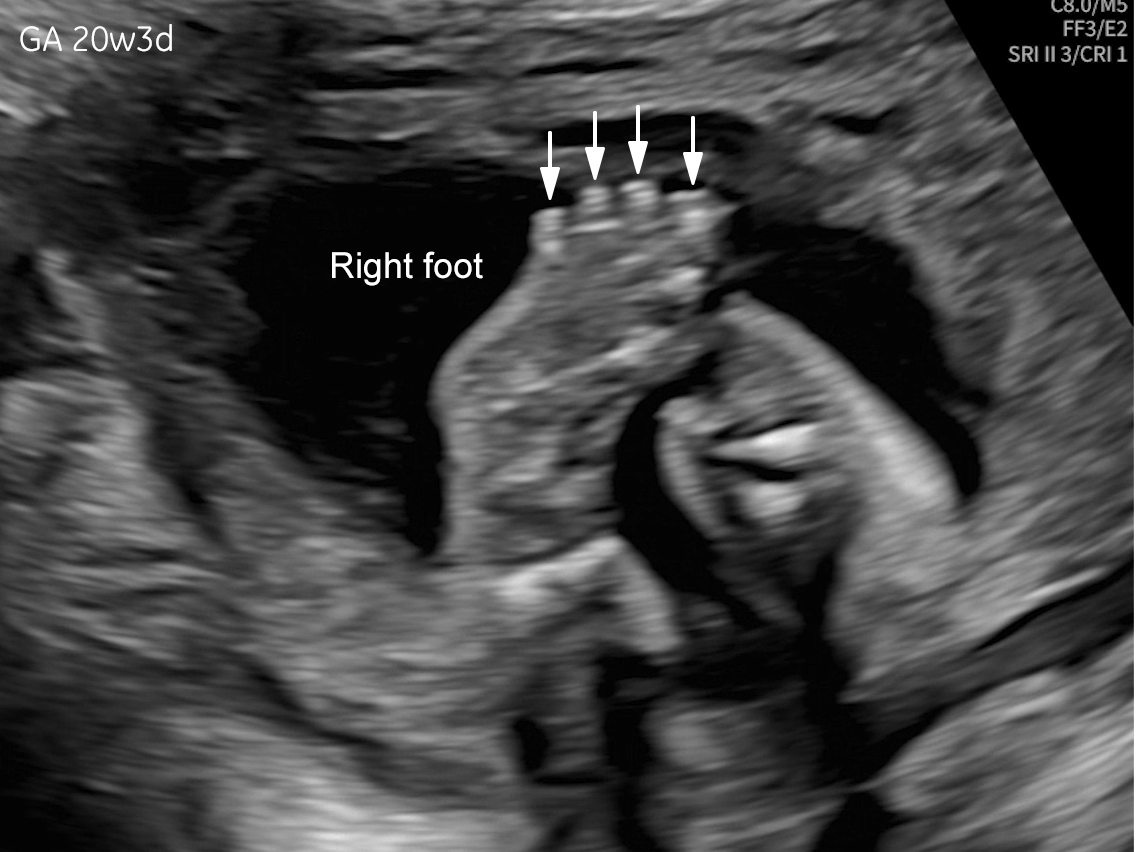
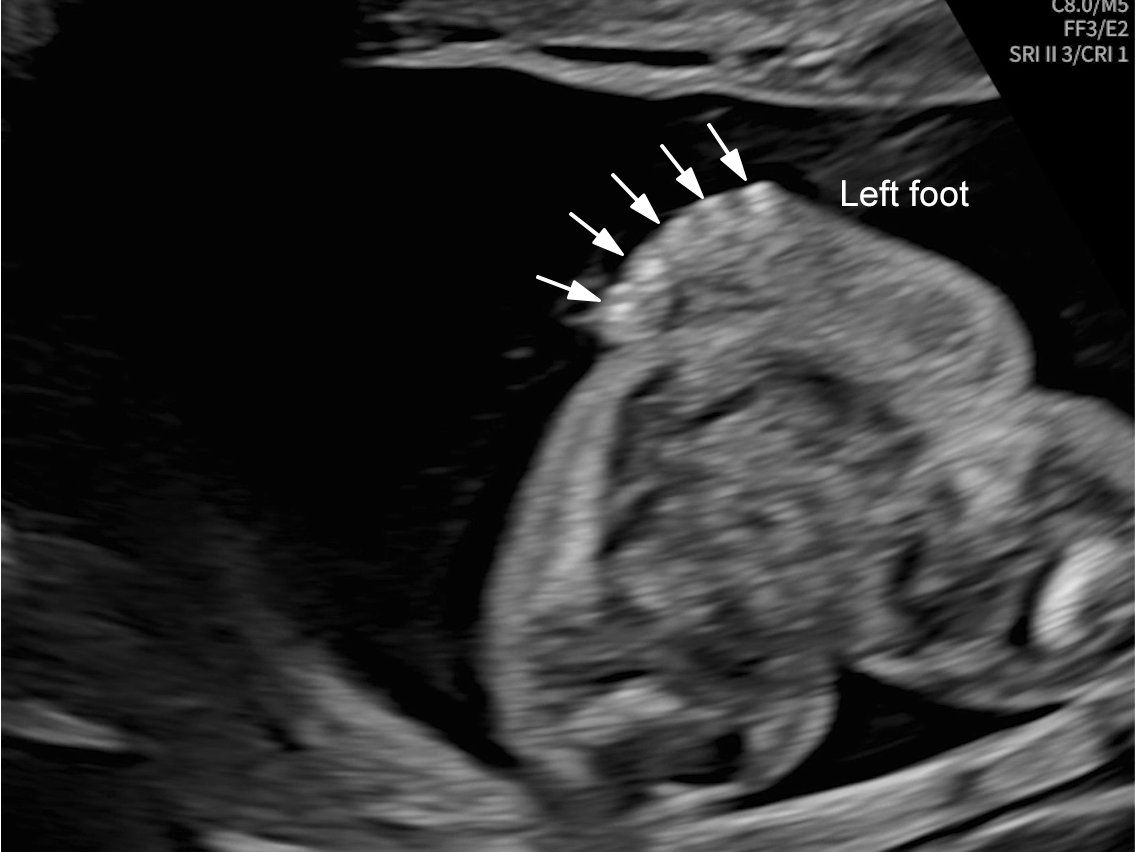
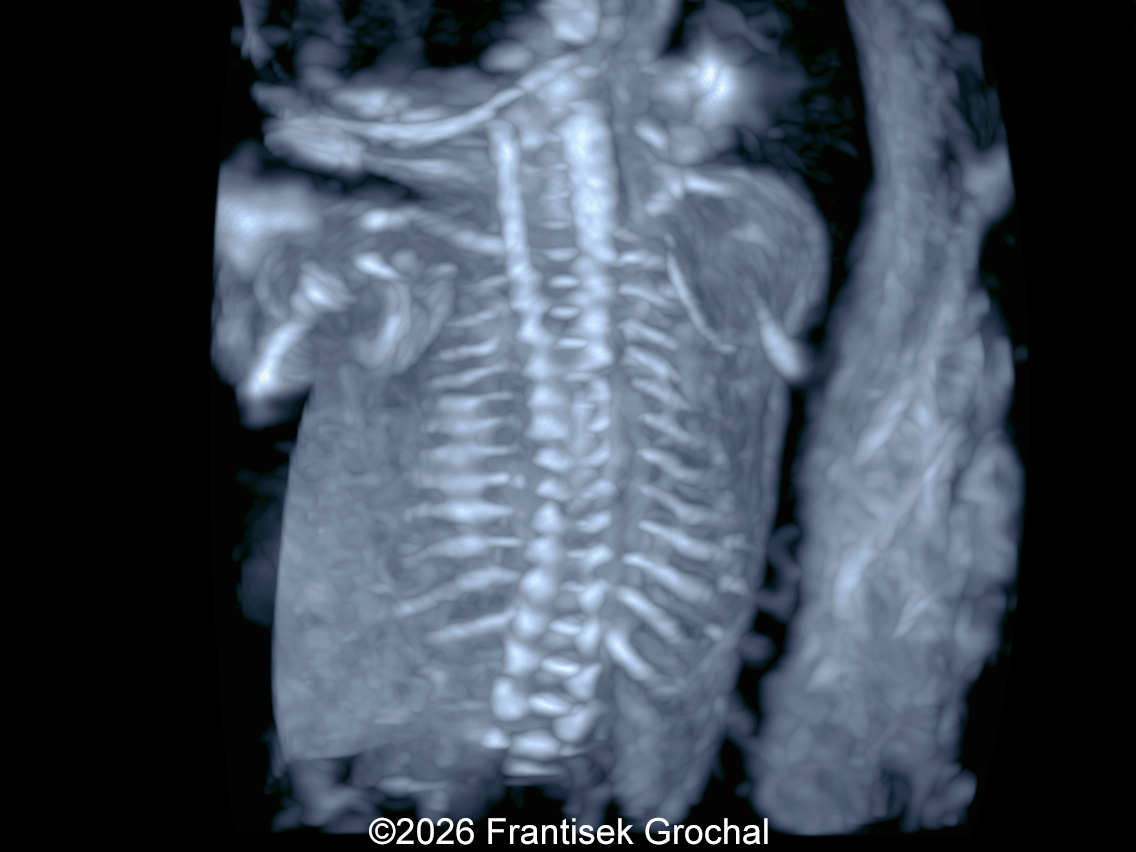
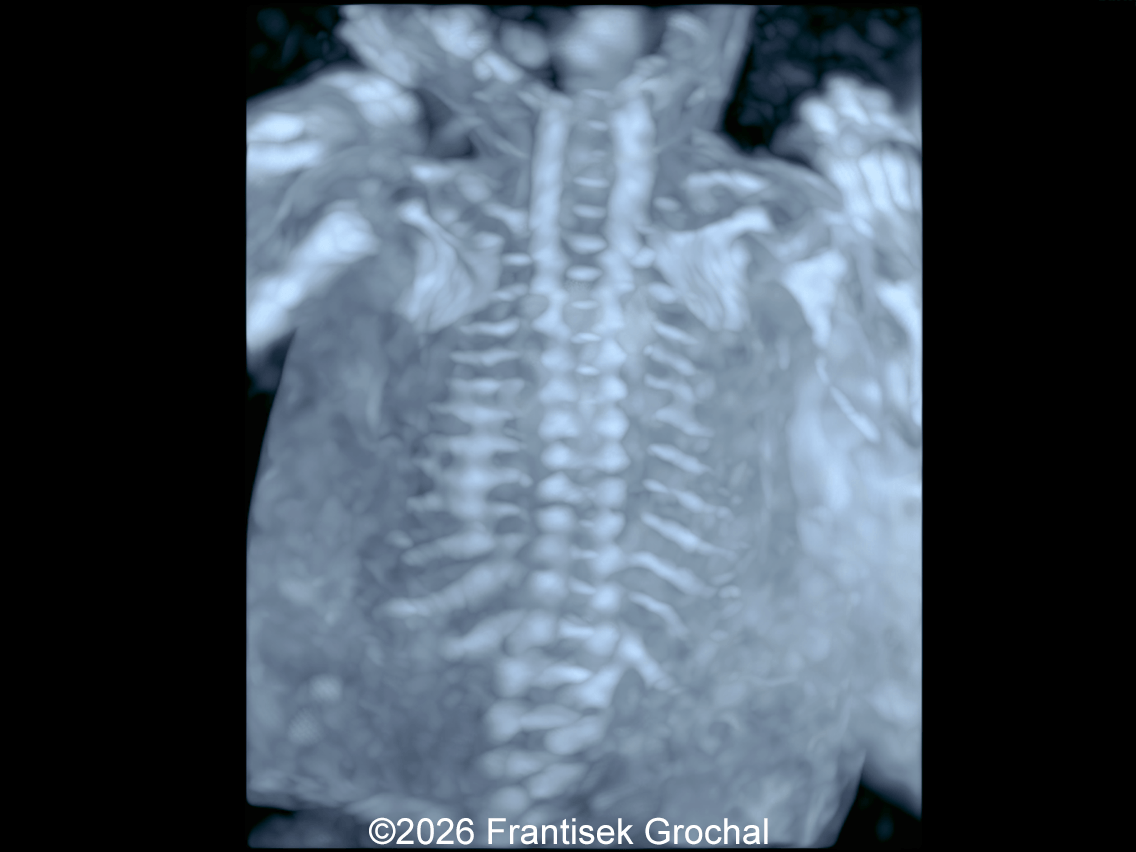
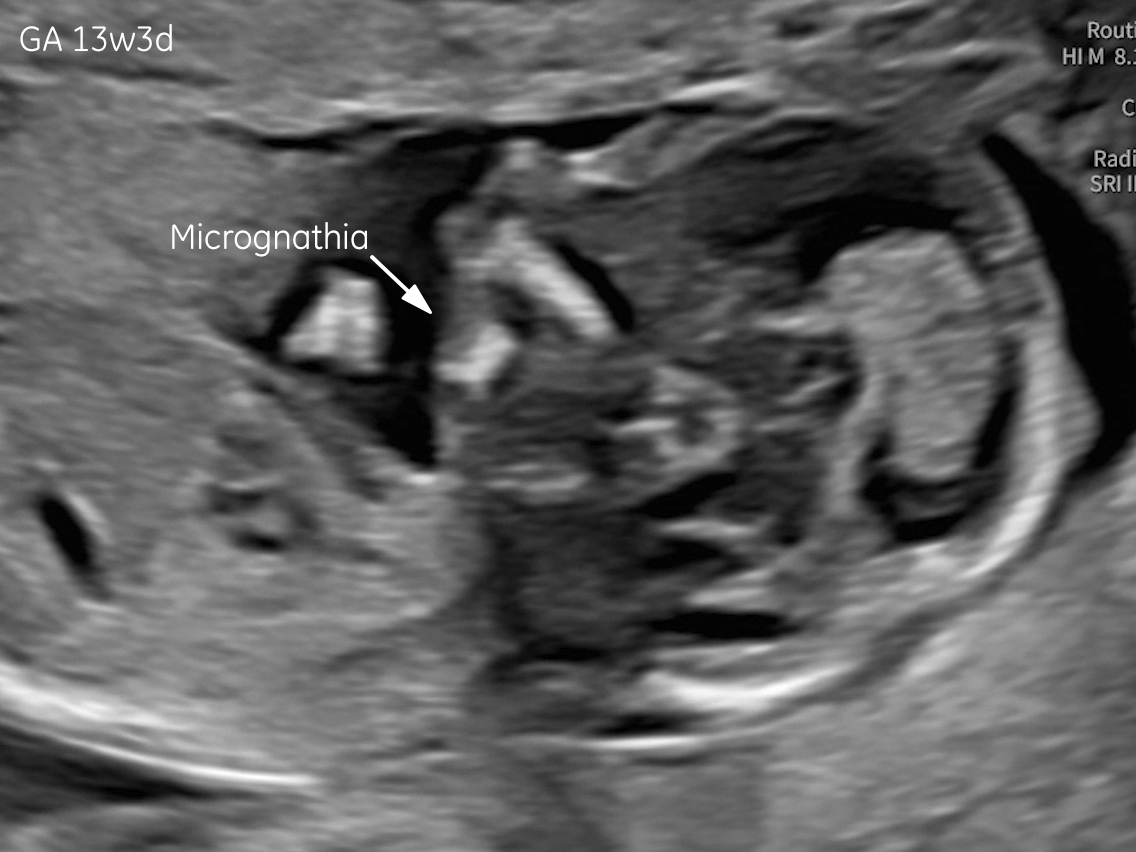
The images and video clips obtained during the pregnancy at 20 weeks gestation demonstrate:
- Micrognathia
- Severe micromelia with deformities of the long bones (angulation and bowing), associated with fixed flexion contractures of the elbows and knees (arthrogryposis)
- Mild hypo-ossification of the cranial bones with normal ossification of the spine
- Right foot oligodactyly (only four toes present)
- Ribs without obvious fractures and no significant thoracic hypoplasia
- Normally developed scapulae
These findings raised suspicion of a lethal skeletal dysplasia. Osteogenesis imperfecta was strongly considered in the differential diagnosis. Amniocentesis identified a pathogenic homozygous variant in the PLOD2 gene (c.1318C>T; p.Arg440Ter). Biallelic pathogenic variants in PLOD2 are associated with Bruck syndrome type 2, an autosomal recessive disorder representing a rare form of osteogenesis imperfecta. After genetic counseling, the parents opted for termination of pregnancy.
Two years later, the patient returned during a subsequent pregnancy at 13 weeks of gestation. The fetus again demonstrated features typical of Bruck syndrome, including severe micromelia, angulated (bowed) long bones with fixed joint contractures of the elbows and knees (arthrogryposis), and micrognathia. Amniocentesis was performed and genetic testing confirmed the same diagnosis as in the previous pregnancy. Given the poor prognosis, the parents again decided to terminate the pregnancy.
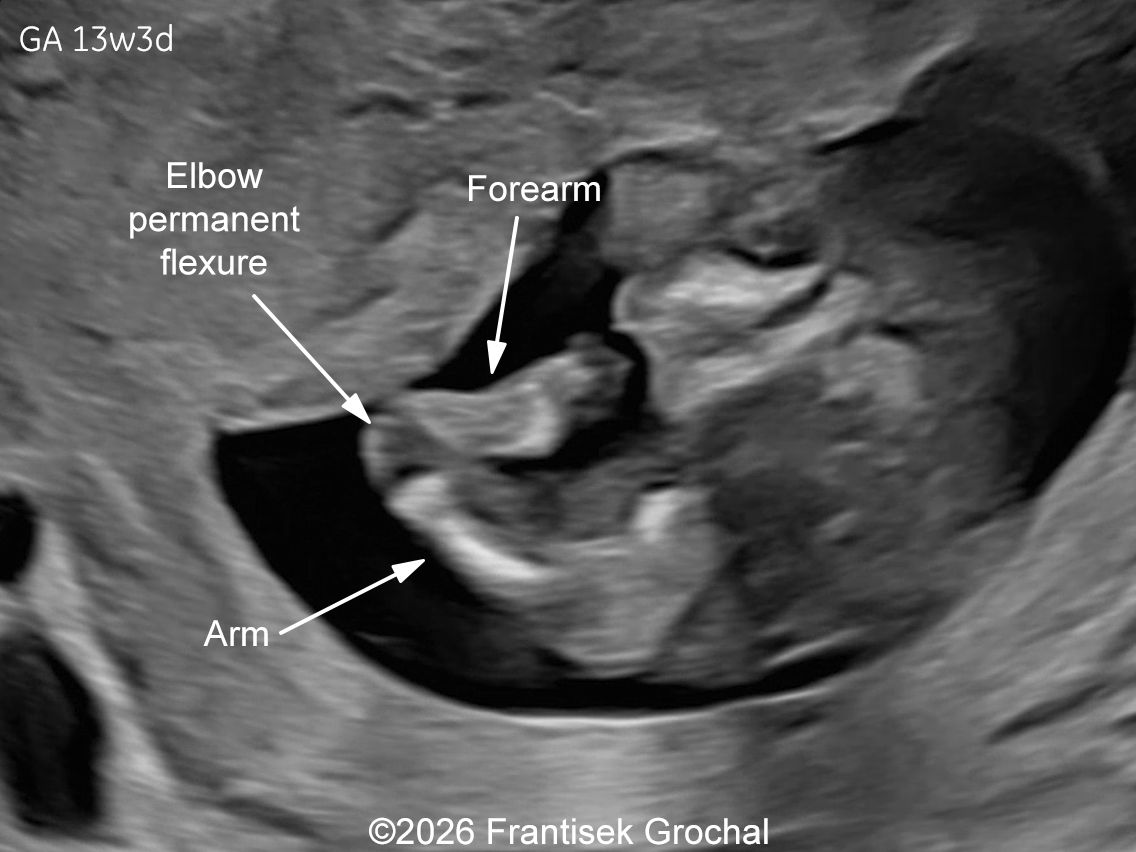
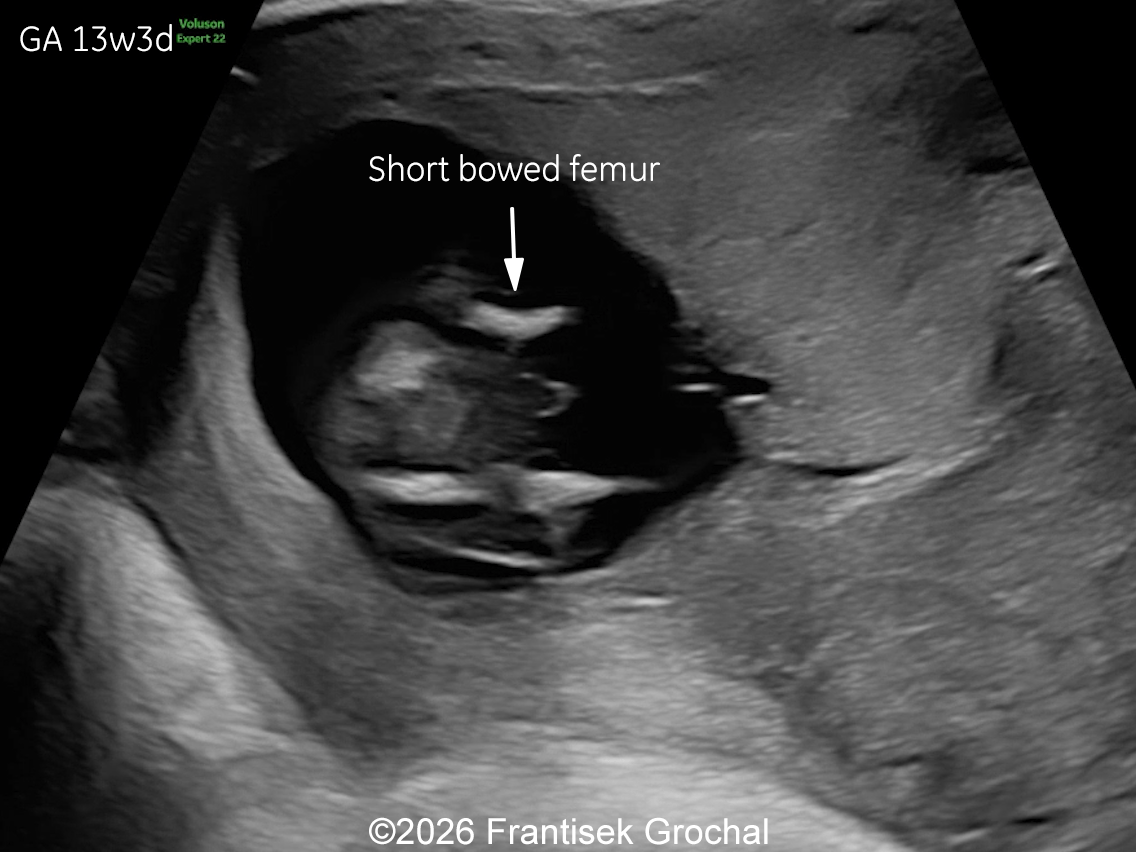
Discussion
The discovery of the first gene responsible for recessive osteogenesis imperfecta in 2006 transformed the understanding of collagen‑related bone dysplasias. Although most cases of osteogenesis imperfecta result from dominant mutations in COL1A1 and COL1A2, the identification of additional genes involved in collagen modification, folding, and trafficking expanded the spectrum of recessive forms [1]. As these genetic subtypes accumulated, Sillence’s 1979 clinical phenotypic classification [2] evolved into a functional genetic classification that groups osteogenesis imperfecta according to the disrupted intracellular or extracellular metabolic pathway [3]:
- Group A: primary defects in collagen structure or processing
- Group B: defects in collagen post‑translational modification
- Group C: abnormalities in collagen folding and cross‑linking
- Group D: defects in ossification or mineralisation
- Group E: impaired osteoblast development leading to collagen insufficiency.
Within group C, Bruck syndrome represents a distinct entity characterized by congenital joint contractures and bone fragility, arising from defects in proteins essential for collagen cross‑linking and extracellular matrix stability.
In 1989, Viljoen et al. [4] described five children with congenital, symmetrical contractures of the knees, ankles, and feet, as well as the presence of Wormian bones and fractures resulting from minimal trauma. Because the contractures were evident at birth, all children were initially diagnosed with congenital arthrogryposis, a diagnosis that was later reconsidered once they began to walk and fractures appeared. Given the striking similarity to the case reported by Bruck in a German medical journal in 1897 [5], the authors proposed the term Bruck syndrome to designate this disorder. However, according to Ha‑Vinh et al. [6], the eponym is historically inaccurate, since Bruck’s original patient had osteogenesis imperfecta and developed joint contractures only later in life rather than congenitally.
The incidence of Bruck syndrome remains unknown. Fewer than 50 to 60 cases have been reported worldwide, supporting a prevalence well below 1 per 1,000,000 [7,8]. The disorder is genetically heterogeneous and results from biallelic pathogenic variants in FKBP10 or PLOD2, both of which encode proteins essential for the post‑translational maturation of type I collagen. Bruck syndrome type 1 (OMIM #259450) is caused by pathogenic variants in FKBP10, located on chromosome 17q21. This gene encodes FKBP65, a component of an endoplasmic reticulum chaperone complex that is crucial for the correct folding and stabilization of type I procollagen [9-11]. In contrast, Bruck syndrome type 2 (OMIM #609220) results from pathogenic variants in PLOD2 (chromosome 3q24), which encodes lysyl hydroxylase 2 (LH2). LH2 catalyzes the hydroxylation of lysine residues in the telopeptides of type I collagen, a modification required for the formation of intermolecular crosslinks and the mechanical stabilization of collagen fibrils [6,12-16]. Schwarze et al. [17] have reported that FKBP65 deficiency in patients with FKBP10 mutations in part leads to impaired LH2‑mediated hydroxylation of lysines in type I collagen telopeptides, which helps explain the phenotypic overlap between Bruck I (FKBP10) and Bruck II (PLOD2).
Postnatally, Bruck syndrome is characterized by bone fragility with recurrent fractures, particularly of the ribs and long tubular bones, often beginning in infancy or early childhood. Affected individuals typically present with multiple joint contractures and cutaneous pterygia. The contractures are congenital and associated with pterygia while the fractures are often of postnatal onset. Therefore, the contractures must be considered as a primary abnormality and not a complication of the fractures or the resulting immobilization [18]. Most patients have normal cognitive development and normal birth length, although some exhibit vertebral abnormalities such as platyspondyly, which may lead to short stature, disproportionate growth, and progressive spinal deformities [19,20]. Many reported patients also present with Wormian bones and congenital talipes, whereas blue sclerae are observed in only a minority of cases (approximately 25%). Camptodactyly and progressive scoliosis are common. There is marked inter‑ and intrafamilial variability, and the clinical phenotype alone does not reliably distinguish between Bruck syndrome types 1 and 2 [15, 21].
Prenatally, the condition has been diagnosed in a small number of cases [18,22-26]. It may present with severe micromelia, bowing or angulation of the long bones, fractures, reduced mineralization, and multiple joint contractures. Additional findings may include hypo-ossification of the calvarium, micrognathia and limb anomalies (clubbed feet). Thoracic hypoplasia and pulmonary insufficiency contribute to the poor prognosis in severe cases. Occasionally, abnormalities of the cardiac valvular structures have been reported in a fetus with Bruck syndrome, which underscores the need for ongoing echocardiographic evaluation throughout pregnancy in these patients [26]. In our case, the combination of severe micromelia, femoral bowing, fixed flexion contractures of the elbows and knees, and mild hypo-ossification of the calvarium raised suspicion for a lethal skeletal dysplasia within the osteogenesis imperfecta spectrum. Molecular testing confirmed a homozygous pathogenic PLOD2 variant, establishing the diagnosis of Bruck syndrome type 2. The recurrence of identical findings in two consecutive pregnancies, a pattern reported in several published cases, is consistent with autosomal recessive inheritance, conferring a 25% recurrence risk.
When both bone fragility and congenital joint contractures are identified prenatally, the differential diagnosis must first consider disorders that present these findings separately. Arthrogryposis is a key distinguishing feature that helps differentiate Bruck syndrome from classic forms of osteogenesis imperfecta as true congenital contractures are not characteristic of osteogenesis imperfecta. Although severe arthrogryposis multiplex congenita may lead to secondary skeletal remodeling, bone fragility, fractures, and generalized undermineralization are not expected. Campomelic dysplasia may show long‑bone bowing and limb positional abnormalities, but fractures and generalized osteopenia are uncommon, and contractures are usually less pronounced. Additional features such as characteristic facial morphology, hypoplastic scapulae, and possible sex reversal assist in differentiation between Campomelic dysplasia and Bruck syndrome [15]. Other rare bent bone dysplasias (e.g., Stüve‑Wiedemann syndrome, kyphomelic dysplasia) may present with limb bowing or contractures, but they typically lack the combination of multiple fractures, undermineralization, and generalized bone fragility that defines Bruck syndrome.
This case highlights the importance of early first and second trimester recognition of skeletal shortening combined with arthrogryposis, which should prompt targeted genetic testing and counselling. Early diagnosis enables informed parental decision-making, including the possibility of legal termination of pregnancy, and allows considerations of options such as early invasive testing or preimplantation genetic testing in future pregnancies.
References
- Forlino A, Cabral WA, Barnes AM, Marini JC. New perspectives on osteogenesis imperfecta. Nat Rev Endocrinol. 2011 Jun 14;7(9):540-557.
- Sillence DO, Senn A, Danks DM. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet. 1979 Apr;16(2):101-116.
- Forlino A, Marini JC. Osteogenesis imperfecta. Lancet. 2016 Apr 16;387(10028):1657-1671.
- Viljoen D, Versfeld G, Beighton P. Osteogenesis imperfecta with congenital joint contractures (Bruck syndrome). Clin Genet. 1989 Aug;36(2):122-126.
- Bruck, A. Multiple fractures associated with joint ankylosis and muscle atrophy. Dtsch. Med Wochenschr. 1897;23:152-155.
- Ha-Vinh R, Alanay Y, Bank RA, et al. Phenotypic and molecular characterization of Bruck syndrome (osteogenesis imperfecta with contractures of the large joints) caused by a recessive mutation in PLOD2. Am J Med Genet A. 2004 Dec 1;131(2):115-120.
- Orphanet. Bruck syndrome [Internet]. Paris: Orphanet; 2024 [cited 2026 Mar 8]. Available from: https://www.orpha.net/en/disease/detail/2771
- Manohar S, Jakes A, Watt-Coote I, Khalil A. Bruck syndrome in pregnancy. BMJ Case Rep. 2024 Sep 10;17(9):e257696.
- Kelley BP, Malfait F, Bonafe L, et al. Mutations in FKBP10 cause recessive osteogenesis imperfecta and Bruck syndrome. J Bone Miner Res. 2011 Mar;26(3):666-672.
- Shaheen R, Al-Owain M, Faqeih E, et al. Mutations in FKBP10 cause both Bruck syndrome and isolated osteogenesis imperfecta in humans. Am J Med Genet A. 2011 Jun;155A(6):1448-1452.
- Moravej H, Karamifar H, Karamizadeh Z, et al. Bruck syndrome - a rare syndrome of bone fragility and joint contracture and novel homozygous FKBP10 mutation. Endokrynol Pol. 2015;66(2):170-174.
- Bank RA, Robins SP, Wijmenga C, et al. Defective collagen crosslinking in bone, but not in ligament or cartilage, in Bruck syndrome: indications for a bone-specific telopeptide lysyl hydroxylase on chromosome 17. Proc Natl Acad Sci U S A. 1999 Feb 2;96(3):1054-1058.
- van der Slot AJ, Zuurmond AM, Bardoel AF, et al. Identification of PLOD2 as telopeptide lysyl hydroxylase, an important enzyme in fibrosis. J Biol Chem. 2003 Oct 17;278(42):40967-40972.
- Puig-Hervás MT, Temtamy S, Aglan M, et al. Mutations in PLOD2 cause autosomal-recessive connective tissue disorders within the Bruck syndrome--osteogenesis imperfecta phenotypic spectrum. Hum Mutat. 2012 Oct;33(10):1444-1449.
- Leal GF, Nishimura G, Voss U, et al. Expanding the Clinical Spectrum of Phenotypes Caused by Pathogenic Variants in PLOD2. J Bone Miner Res. 2018 Apr;33(4):753-760.
- Lv F, Xu X, Song Y, et al. Novel Mutations in PLOD2 Cause Rare Bruck Syndrome. Calcif Tissue Int. 2018 Mar;102(3):296-309.
- Schwarze U, Cundy T, Pyott SM, et al. Mutations in FKBP10, which result in Bruck syndrome and recessive forms of osteogenesis imperfecta, inhibit the hydroxylation of telopeptide lysines in bone collagen. Hum Mol Genet. 2013 Jan 1;22(1):1-17.
- Cuillier F, Alessandri JL, Lemaire P, et al. Bruck syndrome: second antenatal diagnosis. Fetal Diagn Ther. 2007;22(1):23-28.
- Otaify GA, Abdel-Hamid MS, Hassib NF, et al. Bruck syndrome in 13 new patients: Identification of five novel FKBP10 and PLOD2 variants and further expansion of the phenotypic spectrum. Am J Med Genet A. 2022 Jun;188(6):1815-1825.
- Bolshakova OI, Latypova EM, Komissarov AE, et al. Cellular and Molecular Effects of the Bruck Syndrome-Associated Mutation in the PLOD2 Gene. Int J Mol Sci. 2024 Dec 13;25(24):13379.
- Luce L, Casale M, Waldron S. A Rare Case of Bruck Syndrome Type 2 in Siblings With Broad Phenotypic Variability. Ochsner J. 2020 Summer;20(2):204-208.
- Berg C, Geipel A, Noack F, et al. Prenatal diagnosis of Bruck syndrome. Prenat Diagn. 2005 Jul;25(7):535-538.
- Quesada Segura GE, Cantos García C, Lobo Valentín R, et al. Diagnóstico prenatal de síndrome de Bruck en una paciente con tres gestaciones consecutivas afectas. Prog Obstet Ginecol. 2017;60(2):126‑129.
- Zhang J, Hu H, Mu W, et al. Case Report: Exome Sequencing Identified a Novel Compound Heterozygous Variation in PLOD2 Causing Bruck Syndrome Type 2. Front Genet. 2021 Feb 16;12:619948.
- Parvin S, Dhali A, Biswas DN, Ray S. Bruck syndrome: a rare cause of reduced fetal movements. BMJ Case Rep. 2021 Dec 1;14(12):e246786.
- Tran CT, Smet ME, Forsey J, et al. Bruck Syndrome: Beyond the Obvious. Fetal Diagn Ther. 2022;49(11-12):479-485.
Discussion Board
We appreciate your patience as we review all submitted answers. Check back soon to see if you were correct!