Case of the Week #617
(1) Polish Mother's Research Institute - Dept. of Genetics; (2) UCSF Health, San Francisco, CA, United States
A primigravida presented at 19 weeks with initially non-contributive anamnesis. After additional questioning, it was revealed that her brother had schizophrenia, and another maternal male relative was affected with an undiagnosed neurologic condition.
View the Answer Hide the Answer
Answer
We present a case of X-linked hydrocephalus due to L1CAM mutation.
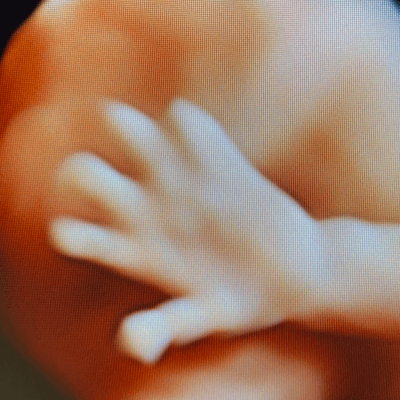
Our imaging revealed dilated lateral and third ventricles, prominent cavum veli interpositi, short dysplastic corpus callosum, and adducted thumbs in a male fetus. Prominent cavum veli interpositi and adducted thumbs are typical findings, however, adducted thumbs are not a constant feature of L1 syndrome, at times visible only temporarily, and not necessarily throughout the whole prenatal period. Sanger sequencing of L1CAM gene, prompted by characteristic phenotype, revealed causative mutation.
Discussion
Congenital hydrocephalus is the abnormal accumulation of intracranial cerebrospinal fluid and occurs in approximately 4.65 per 10,000 births [1]. In 1949, Bickers and Adams identified a family with six male relatives who presented with congenital hydrocephalus related to stenosis of the aqueduct of Sylvius, and hypothesized an x-linked inheritance [2]. X‐linked hydrocephalus is characterized by enlargement of both third and lateral ventricles, agenesis of the corpus callosum, atrophy of the corticospinal descending pathways in the pons and medulla, spasticity of primarily the lower limbs, mental retardation and bilateral adducted thumbs [3]. It affects approximately 0.02-0.04 in 1,000 male births [4]. In 1992, Rosenthal et al first reported a mutation in the L1CAM gene associated with X-linked hydrocephalus [5]. Since then, the association between X-linked hydrocephalus and mutations in the L1CAM gene has been well established.
Neural protein L1 cell adhesion molecule (L1CAM) is an axonal glycoprotein belonging to the immunoglobulin superfamily (IgSF) and plays a key role in neuronal adhesion, neuronal migration, growth cone morphology, neurite outgrowth and myelination [6-8]. Mutation of L1CAM can lead to “L1 syndrome,” which is an X-linked recessive disorder and includes several phenotypes dominated by hydrocephalus [9]. It is also known as CRASH syndrome, an acronym for Corpus callosum hypoplasia, Retardation, Adducted thumbs, Spasticity and Hydrocephalus [10]. L1 syndrome encompasses HSAS (Hydrocephalus with Stenosis of the Aqueduct of Sylvius, OMIM #307000), MASA syndrome (Mental retardation, Aphasia, Spastic paraplegia, Adducted thumbs, OMIM #303350), SPG1 (hereditary complicated spastic paraplegia type 1, OMIM #312900), and x-linked agenesis of the corpus callosum (OMIM #217990) [3,9]. More than 300 pathogenic L1CAM variants have been reported in the Human Gene Mutation Database [11]. Yamasaki et al described the phenotype-genotype correlation between L1CAM mutations and classified them into three types: Class I are mutations that affect the cytoplasmic domain of L1, Class II are missense point mutations in the extracellular domain of the protein, and Class III are nonsense or frame shift mutations that produce a premature stop codon in the extracellular domain of L1 which results in loss of its transmembrane domain and disassociation of L1 with the cell surface. In their study, the authors found that Class III mutations are more likely to produce a severe form of L1 syndrome compared to Class I and II. Class I mutations showed significantly better survival than both Class II and Class III [12]. Vos et al similarly found that children with a truncating mutation were more likely to die before the age of 3 than children with a missense mutation [13].
Several studies have attempted to define which patients should undergo genetic testing for L1CAM mutations. The six typical features of L1 syndrome include: male sex, adducted thumbs, hydrocephalus, corpus callosum abnormalities (complete or partial agenesis with or without Probst bundles, corpus callosum hypoplasia), stenosis of the aqueduct of Sylvius, and abnormalities of the corticospinal tract. Finckh et al reviewed 153 cases of suspected X-linked hydrocephalus and found that 30% had L1CAM mutations causing disease. These mutations were found in 74% of cases with two or more affected family members, and in 63% of cases with three or more L1 syndrome features. Detection rates were increased to 93% in patients with three or more familial cases, and two or more L1 syndrome traits [14]. Vos et al evaluated 367 patients with suspected L1 syndrome and identified 20% with disease-causing mutations in the L1CAM gene. In patients with a positive family history of hydrocephalus, and three or more clinical characteristics of L1 syndrome, 85% were found to have mutations in the L1CAM gene [13]. Adle-Biasset et al reviewed 138 cases of suspected X-linked hydrocephalus and found L1CAM mutations in 41%. They reported the sensitivity, specificity and positive predictive value of the six L1 syndrome signs and found that each was associated with high sensitivity (88-100 %) but low specificity (10-54%), except for adducted thumbs with a specificity of 73%. In patients presenting with five or more L1 syndrome features, mutations in L1CAM were detected in 80% [15]. In conclusion, screening for L1CAM should be considered in male fetuses with a family history of congenital hydrocephalus and at least two characteristics of L1 syndrome. Our case was a male fetus with three of the classic findings for L1 syndrome and family history of an unknown neurologic condition which prompted L1CAM testing.
The differential diagnosis for L1 syndrome includes fetal alcohol syndrome and aqueductal atresia spectrum. Patients with fetal alcohol syndrome have craniofacial dysmorphism and a maternal history of alcoholism, which are lacking in L1 syndrome. Aqueductal atresia spectrum consists of atresia-forking of the aqueduct of Sylvius, often associated with a fusion of the colliculi, rhombencephalosynapsis, diencephalosynapsis and corpus callosum agenesis [15].
References
[1] Garne E, Loane M, Addor MC, et al. Congenital hydrocephalus--prevalence, prenatal diagnosis and outcome of pregnancy in four European regions. Eur J Paediatr Neurol. 2010 Mar;14(2):150-5.
[2] Bickers DS, Adams RD. Hereditary stenosis of the aqueduct of Sylvius as a cause of congenital hydrocephalus. Brain. 1949 Jun;72(Pt. 2):246-62.
[3] Kong W, Wang X, Zhao J, et al. A new frameshift mutation in L1CAM producing X‐linked hydrocephalus. Mol Genet Genomic Med. 2019 Nov 22;8(1):e1031.
[4] Halliday J, Chow CW, Wallace D, et al. X linked hydrocephalus: a survey of a 20 year period in Victoria, Australia. J Med Genet. 1986 Feb;23(1):23–31.
[5] Rosenthal A, Jouet M, Kenwrick S. Aberrant splicing of neural cell adhesion molecule L1 mRNA in a family with X-linked hydrocephalus. Nat Genet. 1992 Oct;2(2):107-12.
[6] Walsh FS, Meiri K, Doherty P. Cell signaling and CAM-mediated neurite outgrowth. Soc Gen Physiol Ser. 1997:52:221-6.
[7] Barbin G, Aigrot MS, Charles P, et al. Axonal cell-adhesion molecule L1 in CNS myelination. Neuron Glia Biol. 2004 Feb;1(1):65-72.
[8] Schmid RS, Maness PF. L1 and NCAM adhesion molecules as signaling co-receptors in neuronal migration and process outgrowth. Curr Opin Neurobiol. 2008 Jun;18(3):245–250.
[9] Ochando I, Vidal V, Gascón J, et al. Prenatal diagnosis of X-linked hydrocephalus in a family with a novel mutation in L1CAM gene. J Obstet Gynaecol. 2016;36(3):403-5.
[10] Fransen 1E Lemmon V, Van Camp G, et al. CRASH syndrome: clinical spectrum of corpus callosum hypoplasia, retardation, adducted thumbs, spastic paraparesis and hydrocephalus due to mutations in one single gene, L1. Eur J Hum Genet. 1995;3(5):273-84.
[11] Šubrt I, Zavoral T, Strych L, et al. A recurrent synonymous L1CAM variant in a fetus with hydrocephalus. Hum Genome Var. 2024 Jan 23;11:4.
[12] Yamasaki M, Thompson P, Lemmon V. CRASH Syndrome: Mutations in L1CAM Correlate with Severity of the Disease. Neuropediatrics. 1997 Jun;28(3):175–178.
[13] Vos YJ, de Walle HEK, Bos KK, et al. Genotype-phenotype correlations in L1 syndrome: a guide for genetic counselling and mutation analysis. J Med Genet. 2010 Mar;47(3):169-75.
[14] Finckh U, Schröder J, Ressler B, et al. Spectrum and detection rate of L1CAM mutations in isolated and familial cases with clinically suspected L1-disease. Am J Med Genet. 2000 May 1;92(1):40-6.
[15] Adle-Biassette H, Saugier-Veber P, Fallet-Bianco C, et al. Neuropathological review of 138 cases genetically tested for X-linked hydrocephalus: evidence for closely related clinical entities of unknown molecular bases. Acta Neuropathol. 2013 Sep;126(3):427-42.
Discussion Board
Winners

Javier Cortejoso Spain Physician
paola quaresima Italy Physician

belen garrido Spain Physician

Andrii Averianov Ukraine Physician

Ana Ferrero Spain Physician

Mayank Chowdhury India Physician

Ivan Ivanov Russian Federation Physician

Boujemaa Oueslati Tunisia Physician

Aysegul Ozel Turkey Physician

Caroline Reichert Garcia Brazil Physician
Irvin Jacob Vélez Machorro Mexico Physician
Olivia Ionescu United Kingdom Physician
Marianovella Narcisi Italy Physician

Elena Andreeva Russian Federation Physician
Muradiye YILDIRIM Turkey Physician

ALBANA CEREKJA Italy Physician

Deval Shah India Physician

Murat Cagan Turkey Physician

Sonio Sonio France AI
gholamreza azizi Iran, Islamic Republic of Physician

Ionut Valcea Romania Physician
Đặng Mai Quỳnh Viet Nam Physician

Kareem Haloub Australia Physician
Martina Vagaská Slovakia Physician

Annette Reuss Germany Physician

shay kevorkian Israel Physician

Ismail Guzelmansur Turkey Physician

Sruthi Pydi India Physician
Shina Kaur India Physician
Pallavi Mishra Kenya Physician

Eylem Eşsizoğlu Turkey Physician

Nguyễn Lê Hoàng Viet Nam Physician

Denys Saitarly Israel Physician

Tetiana Ishchenko Ukraine Physician
Eduardo Namura Di Thomaz Brazil Physician

Hana Habanova Slovakia Physician

Navya Sri Mopada India Physician

Jagdish Suthar India Physician
Akhani Madhvi India Physician

Fco Javier Martínez Cortés Spain Physician
Viktoriia Putsenko Russian Federation Physician