Case of the Week #576
(1) Russia; (2) Australia
Case Report: 24-year-old primigravida presented for ultrasound examination at 20-21 weeks of pregnancy. There was no family history of hereditary disease.
View the Answer Hide the Answer
Answer
We present a mid-trimester diagnosis of the congenital lissencephaly with dysgenesis of corporis collosum
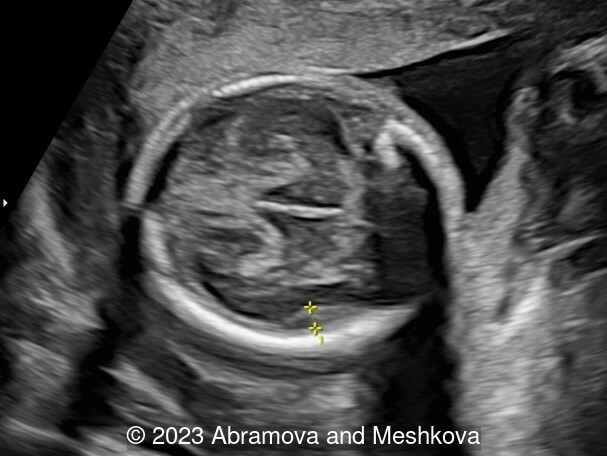
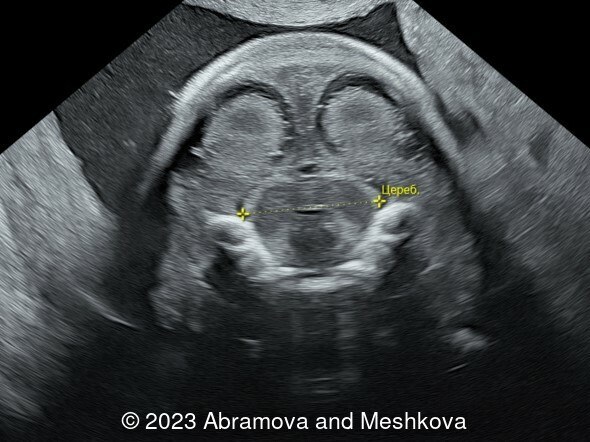
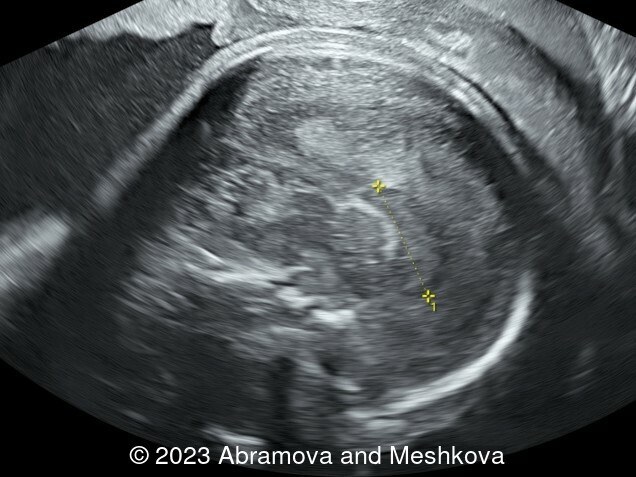
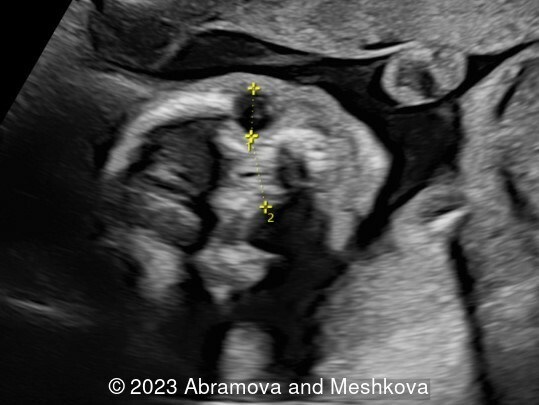
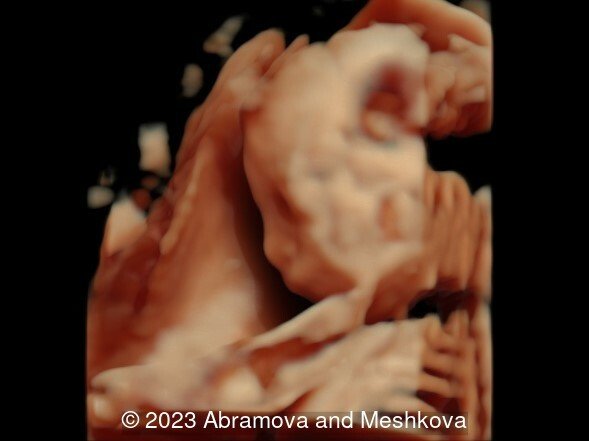
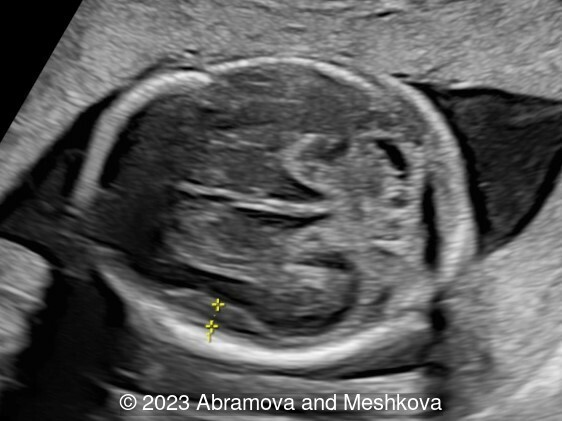
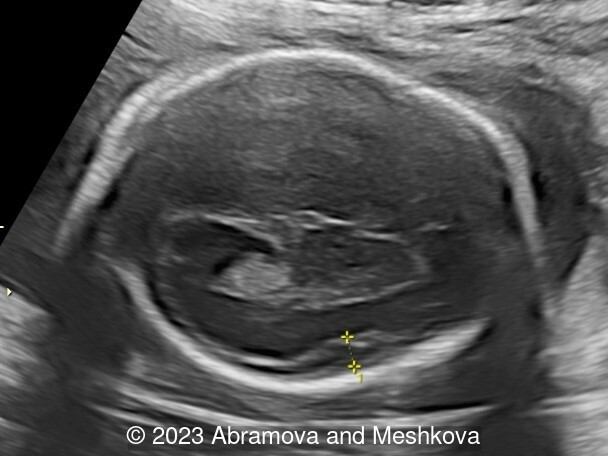
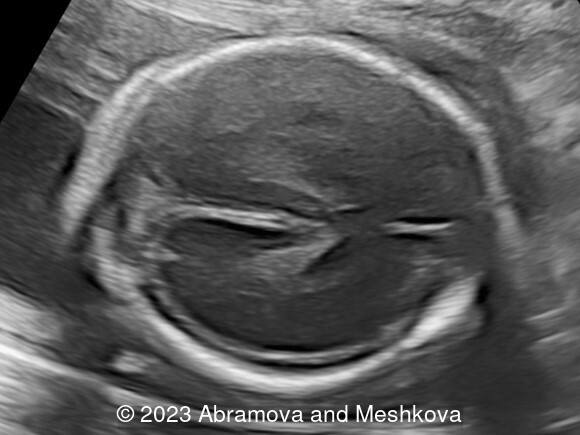
Our ultrasound at 20-21 weeks found the following: an absence of cavum pellucidum, smooth Silvia sulci and absence of temporal-occipital sulcation, abnormal corporis callosum, facial dysmorphism (hypertelorism, flat profile). The next ultrasound examination a week later revealed the same features and MRI was recommended, which agreed with ultrasound findings. The followed ultrasound at 23-24 weeks of gestation confirmed the diagnosis of lissencephaly, agenesis of cavum pellucidum and hypoplasia of the vermis of cerebellum.
A genetic consultation was recommended. The couple decided to terminate the pregnancy. The exome sequencing did not revealed pathogenic variants related to the fetal phenotype, however, a heterozygous variant was identified in the nucleotide sequence in the gene LAMB1(7-107936661-G-C), with aa amino acid substitution at 1331 protein position (Ala1331Gly, NM_002291, rs 201636713). A homozygous and compound-heterozygous variant in LAMB-1 has been described among patients with lissencephaly (OMIM:615191). Lissencephaly 5 (LIS5) is caused by homozygous or compound heterozygous mutation in the LAMB1 gene (150240) on chromosome 7q31. However, according to existing information, this mutation is considered to have uncertain clinical significance.
Discussion
Lissencephaly (smooth-brain, LIS) is a spectrum of severe brain malformations caused by the failure of migrating neurons to reach optimal positions in the developing cerebral cortex, resulting in broad or absent cerebral convolutions or gyri, abnormally thick cortex and histopathologic evidence of abnormal cortical structure. Most types of lissencephaly result from incomplete neuronal migration to the cortex during the third and fourth months of gestation. Classical lissencephaly or ‘smooth brain’ is a severe malformation of the brain manifest by a smooth cerebral surface with lesser involvement of the cerebellum and other rhombic lip derivatives. It results from incomplete neuronal migration to the cerebral cortex and rhombic lip derivatives at 9–13 weeks of embryonic development [1] Macroscopically, this produces marked widening of the cortical gyri and a reduction in the number and complexity of sulci, producing abnormally smooth cerebral hemispheres. Microscopically, there is often disruption of the normal six-layer structure of the neocortex. Affected children have severe or profound mental retardation, epilepsy, and subtle facial abnormalities especially bitemporal hollowing and small jaw [2].
Lissencephaly encompasses a spectrum of agyral malformations from complete agyria (absent gyri) to regional pachygyria (broad gyri), and merges with subcortical band heterotopia. LIS is always associated with an abnormally thick cortex, reduced or abnormal lamination and diffuse neuronal heterotopia [3]. SBH or ‘double cortex’ consists of circumferential bands of heterotopic neurons located just beneath the cortex and separated from it by a thin band of white matter [4,5].
Agyria was defined as cortical regions with sulci >3 cm apart, and pachygyria as abnormally wide gyri with sulci 1.5-3 cm apart. Based on the cortical thickness on MRI images, agyria and pachygyria can generally be divided into two groups, classic LIS with cortex ~10-15 mm thick and several variant LIS subtypes with cortex usually 5-10 mm thick. SBH presents as longitudinal bands of gray matter separated from the cortex by a zone of white matter and separated from the lateral ventricles by another zone of white matter [6].
Classification
Classical lissencephaly occurs in several malformation syndromes. Miller–Dieker syndrome (MDS) consists of classical lissencephaly, characteristic facial abnormalities and sometimes other birth defects [7]. The facial changes consist of prominent forehead, bitemporal hollowing, short nose with upturned nares, flat midface, protuberant upper lip with thin vermillion border and small jaw. It is associated with visible or submicroscopic rearrangements within chromosome band 17p13.3 in almost all patients [8].
Isolated lissencephaly sequence (ILS) consists of classical lissencephaly and its direct sequela with no other major anomalies [2]. Submicroscopic deletions of chromosome 17p13.3 have been detected in almost 40% of patients [8]. Classical lissencephaly also occurs in X-linked lissencephaly and subcortical band heterotopia, which has been mapped to chromosome Xq22.3–q23 [4].
The most common type, known as classical lissencephaly (previously type I), has a very thick 10–20 mm cortex and no other major brain malformations. This is the only type associated with subcortical band heterotopia. Less common types are associated with agenesis of the corpus callosum or severe cerebellar hypoplasia [9,10].
Etiology
Migration of post-mitotic neurons from the ventricular zone to form the cortical plate comprises one of the most critical stages in brain development. The progress in molecular genetics has led to identification of 31 lissencephaly-associated genes with an overall diagnostic yield of over 80%. The number of the LIS-associated genes has increased rapidly over the past five years [11].
Over 25 syndromes with lissencephaly or other neuronal migration disorders have been described. Among them are syndromes with several different patterns of inheritance including chromosomal or de novo mutation, autosomal dominant, autosomal recessive, X-linked and unknown. Genetic counseling thus differs greatly between syndromes. The genes responsible for several of the lissencephaly syndromes have been mapped. X-linked lissencephaly has tentatively been mapped to chromosome Xq22 based on observation of a single X-autosomal translocation in a girl. Both Miller-Dieker syndrome and isolated lissencephaly sequence have been mapped to chromosome 17p13.3 due to detection of deletions and other structural chromosome rearrangements. Fukuyama congenital muscular dystrophy has been mapped to chromosome 9q31-33 by homozygosity mapping.
Prevalence
An estimated prevalence of lissencephaly is 1 in 100,000 births [12].
Recurrence risk
The inheritance pattern of these genes is autosomal dominant (LIS1—chromosome 17) and X-linked (DCX). However, the majority of cases are a result of de novo mutations. In a small number of cases, there may be an identifiable parental genetic defect. For example, in cases where a female with a mild phenotype carries a defective copy of the DCX gene, the recurrence risk may be as high as 50%. Alternatively, when one parent has a balanced translocation involving the LIS1 gene, the recurrence risk for isolated lissencephaly sequence is thought to be around 10–15% [13]. The recurrence risk for cobblestone lissencephaly is 25% (autosomal recessive inheritance), as is the case for lissencephaly with cerebellar hypoplasia.
Diagnosis
Prenatal diagnosis of lissencephaly is usually possible only during the third trimester after the 27th gestational week [14]. Extended lissencephaly with cortical thickness >10 mm can reliably be recognized on fetal MRI, and presents as varying degrees of simplification of the gyral pattern as well as occasionally a thick band of arrested neurons within the developing white matter [15]. Several early signs such as ventriculomegaly, delayed opercular development and cavitation, and dysmorphic features of the ganglionic eminence have been described by 20 to 24 gestational weeks [14,15]. However, none of these features are specific for lissencephaly. Careful examination of the sulcation patterns in the second trimester may suggest lissencephaly based on delays in sulcation, but has a high rate of false-positive findings [16 ,17]. Early genome wide genetic testing in at-risk foetuses may help interpret early signs suggesting malformations of cortical development, and lead to the diagnosis before the lissencephaly pattern can be reliably recognized on ultrasound in late pregnancy.
The prenatal diagnosis of lissencephaly is challenging. In utero magnetic resonance imaging may provide additional clinically relevant information compared with ultrasound alone. This is predominantly due to the superior tissue contrast and resolution of MRI when compared with ultrasound, thus it can be valuable in showing fetal brain pathology [18,19]. There are, however, fundamental difficulties in diagnosing abnormalities such as lissencephaly in utero with any imaging modality. The fetal brain normally undergoes significant developmental change in the second and third trimesters, particularly in terms of gyration and sulcation. Most centres do not perform prenatal MRI before 18 weeks, and at that stage, the normal fetal brain is smooth and relatively featureless. Over the next 20 weeks, the major sulci and gyri form in a predictable fashion, producing the highly complex surface features associated with the term fetus [20,21]. Reduced sulcation is a major part of the diagnosis of lissencephaly in children, thus, in the fetus, it is likely that there is a gestational age before which a diagnosis of lissencephaly is not possible with certainty.
Associated anomalies
Duodenal atresia, urinary tract abnormalities, congenital heart defects, cryptorchidism, inguinal hernia, clinodactyly, polydactyly, and ear anomalies may be found.
Prognosis
The prognosis in children affected by lissencephaly is generally poor, with a high likelihood of seizures and severe neurodevelopmental problems. A majority of lissencephaly patients come to medical attention during the first year of life, presenting with various combinations of hypotonia, feeding difficulties, global developmental delay and seizures [22]. However, several recently characterised lissencephaly subtypes have later-onset manifestations. Patients with CRADD or PIDD1-associated lissencephaly may show only mild or even borderline intellectual disability and come to medical attention only after the disorder was diagnosed in another family member [23,24].
Females with DCX-associated thin subcortical band heterotopia may have average cognitive skills or borderline intellectual disability with no other symptoms, while thick subcortical band heterotopia are typically associated with more severe problems. Seizures often begin during the first years of life, but rarely the onset can be delayed until the second decade [25].
Management
Clinical management of patients with lissencephaly remains challenging and still lacks specific therapeutic approaches. The treatment remains symptomatic, depending on the individual clinical manifestation. During the first year of life, the evaluation includes neurologic evaluation, nutritional status, aspiration risk, and developmental assessments. Additional evaluations may include orthopedics and rehabilitation with the assessment of the clubfoot or scoliosis, as well as ophthalmological and audiological evaluations. Early intervention programs may be helpful. With a very few exceptions, epilepsy affects the vast majority of the lissencephaly patients and represents the major clinical challenge as lissencephaly is frequently associated with the drug-resistant epilepsy. Several genotype-phenotype correlations could be made with the first recommendations regarding the choice and combination of the anti-epileptic drugs in patients with LIS1-associated lissencephaly.
References
[1] Dobyns WB. (1987) Developmental aspects of lissencephaly and the lissencephaly syndromes. In Gilbert EF. and Opitz JM. (eds) Genetic aspects of developmental pathology. New York: Alan R. Liss. Birth Defects Orig. Artic. Ser., 23, 225–241.
[2] Dobyns WB, Elias ER, Newlin AC, et al. Causal heterogeneity in isolated lissencephaly. Neurology. 1992 Jul;42(7):1375-88.
[3] Crome, L. (1956) Pachygyria. J. Pathol. Bacteriol., Friede, R.L. (1989) Developmental Neuropathology, 2nd edn. Springer, Berlin 71,335–352.
[4] Dobyns WB, Andermann E, Andermann F, et al. X-linked malformations of neuronal migration. Neurology. 1996 Aug;47(2):331-9.
[5] Barkovich AJ, Guerrini R, Battaglia G, et al. Band heterotopia: correlation of outcome with magnetic resonance imaging parameters. Ann Neurol. 1994 Oct;36(4):609-17.
[6] Oegema R, Barakat TS, Wilke M, et al. International consensus recommendations on the diagnostic work-up for malformations of cortical development. Nat Rev Neurol Nov;16(11):618-635.
[7] Dobyns WB, Curry CJR, Hoyme HE, et al. Clinical and molecular diagnosis of Miller–Dieker syndrome. Am J Hum Genet. 1991 Mar; 48(3): 584–594.
[8] Dobyns WB, Reiner O, Carrozzo R, et al. Lissencephaly, a human brain malformation associated with deletion of the LIS1 gene located at chromosome 17p13. JAMA. 1993 Dec 15;270(23):2838-42.
[9] Dobyns WB, Berry-Kravis E, Havernick NJ, et al. (1999) X-linked lissencephaly with absent corpus callosum and ambiguous genitalia. Am J Med Genet. 1999 Oct 8;86(4):331-7.
[10] Ross ME, Swanson K, Dobyns WB. Lissencephaly with cerebellar hypoplasia (LCH): a heterogeneous group of cortical malformations. Neuropediatrics. 2001 Oct;32(5):256-63.
[11] Donato ND, Timms AE, Aldinger KA et al, Analysis of 17 genes detects mutations in 81% of 811 patients with lissencephaly. Genet Med. 2018 Nov;20(11):1354-1364.
[12] Barkovich AJ, Kuzniecky RI, Jackson GD, et al. Classification system for malformations of cortical development: update 2001. Neurology. 2001 Dec 26;57(12):2168-78.
[13] Guerrini R, Dobyns WB, Barkovich AJ. Abnormal development of the human cerebral cortex: genetics, functional consequences and treatment options. Trends Neurosci. 2008 Mar;31(3):154-62.
[14] Lerman-Sagie T, Leibovitz Z, (2016) Malformations of cortical development: From postnatal to Fetal Imaging. Can J Neurol Sci. 2016 Sep;43(5):611-8.
[15] Choi JJ, Yang E, Soul JS, et al. Fetal magnetic resonance imaging: supratentorial brain malformations. Pediatr Radiol. 2020 Dec;50(13):1934-1947.
[16] Griffiths PD, Severino M, Jarvis D. et al. Changes in appearance of cortical formation abnormalities in the foetus detected on sequential in utero MR imaging. Eur Radiol. 2021 Mar;31(3):1367-1377.
[17] Williams F, Griffiths PD. In utero MR imaging in fetuses at high risk of lissencephaly. Br J Radiol. 2017 Apr;90(1072):20160902.
[18] Griffiths PD, Reeves MJ, Morris JE, et al. A prospective study of fetuses with isolated ventriculomegaly investigated by antenatal ultrasound and in utero MR. AJNR Am J Neuroradiol. 2010 Jan;31(1):106-11.
[19] Whitby EH, Paley MN, Sprigg A, et al. Comparison of ultrasound and magnetic resonance imaging in 100 singleton pregnancies with suspected brain abnormalities. BJOG. 2004 Aug;111(8):784-92.
[20] Levine D, Barnes PD. Cortical maturation in normal and abnormal fetuses as assessed with prenatal MR imaging. Radiology. 1999 Mar;210(3):751-8.
[21] Fogliarini C, Chaumoitre K, Chapon F, et al. Assessment of cortical maturation with prenatal MRI. Part I: normal cortical maturation. Eur Radiol. 2005 Aug;15(8):1671-85.
[22] Dobyns WB. The clinical patterns and molecular genetics of lissencephaly and subcortical band heterotopia. Epilepsia. 2010 Feb;51 Suppl 1:5-9.
[23] Donato ND, Jean YY, Maga AM et al. Mutations in CRADD Result in Reduced Caspase-2-Mediated Neuronal Apoptosis and Cause Megalencephaly with a Rare Lissencephaly Variant. Am J Hum Genet. 2016 Nov 3;99(5):1117-1129.
[24] Harel T, Hacohen N, Shaag A, et al. Homozygous null variant in CRADD, encoding an adaptor protein that mediates apoptosis, is associated with lissencephaly. Am J Med Genet A. 2017 Sep;173(9):2539-2544.
[25] Guerrini R, Moro F, Andermann E, et al. Nonsyndromic mental retardation and cryptogenic epilepsy in women with Doublecortin gene mutations. Ann Neurol. 2003 Jul;54(1):30-7.
Discussion Board
Winners

Javier Cortejoso Spain Physician

Umber Agarwal United Kingdom Maternal Fetal Medicine

Igor Yarchuk United States Sonographer
Larysa Gazarova United States Physician

Andrii Averianov Ukraine Physician

Ana Ferrero Spain Physician

Mayank Chowdhury India Physician

Oskar Sylwestrzak Poland Physician

Vladimir Lemaire United States Physician

Boujemaa Oueslati Tunisia Physician

Tatiana Koipish Belarus Physician

Halil Mesut Turkey Physician
Crismaru Iulia Romania Physician

doğa öcal Turkey Physician
Mária Brešťanská Slovakia Physician

Rasha Abo Almagd Egypt Physician

Inass Osman United Kingdom Physician

Ionut Valcea Romania Physician

Sviatlana Akhramovich Belarus Physician

Prakriti Patil India Physician
Maria Chiara Marra Italy Physician

BHOOMIKA SHARMA India Physician

Kadriye yakut yücel Turkey Physician

Nguyen Xuan Cong Viet Nam Physician