Case of the Week #554
Vinmec Times City Hospital, Vietnam
Posting Dates: March 1 - March 14, 2022
Case Report: A healthy 30-year-old G2P1 with previous uneventful pregnancy was referred to our center at 31 gestational weeks because of suspected abnormalities on prenatal ultrasound. First trimester screening had shown low risk combined test with nuchal translucency of 1mm. There was no consanguinity or any other relevant past history.
Fetal biometry showed the following:
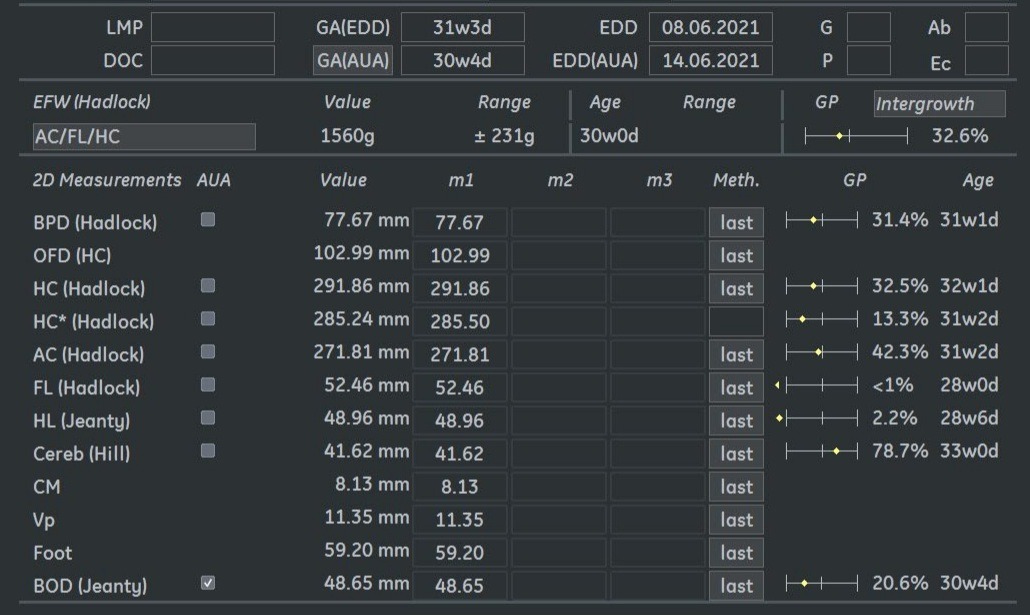
Ultrasound examination at 31 weeks showed multiple abnormalities below:
View the Answer Hide the Answer
Answer
In this report, we present a case of Zellweger spectrum disorder with associated prenatal ultrasound soft markers and clinical exome sequencing confirmation.
Ultrasound at 23 gestational weeks at a local healthcare center revealed rhizomelia and mild bilateral ventriculomegaly. Amniocentesis was performed but found no abnormality by chromosomal microarray analysis.
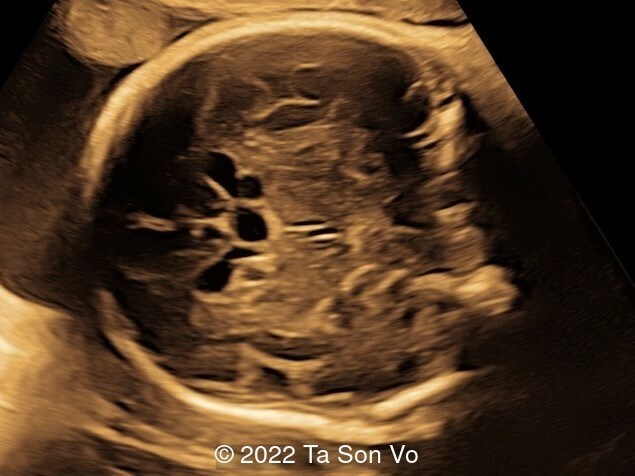
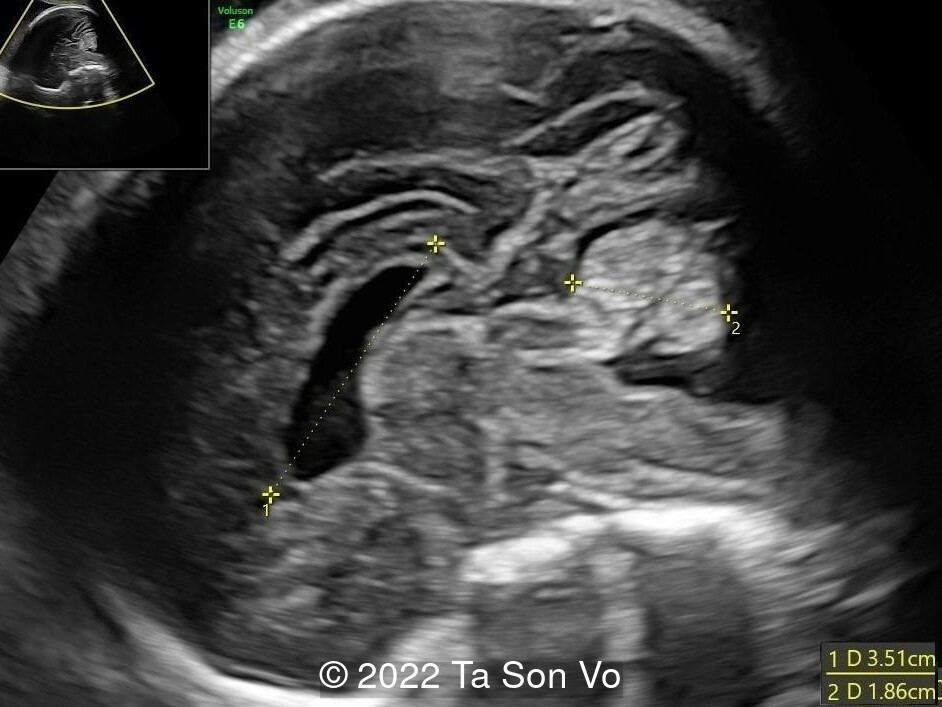
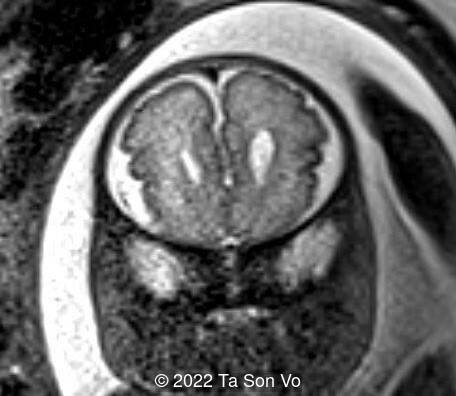
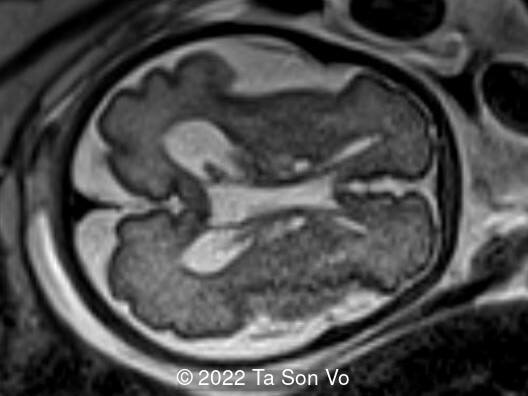
At 31 gestational weeks, a level 3 ultrasound examination in our center showed mild right-sided ventriculomegaly, subependymal cysts, thin but morphologically normal corpus callosum, Blake’s pouch cyst with abnormal bullet shape of 4th ventricle, hepatosplenomegaly and rhizomelia with stippled epiphyses. Additionally, magnetic resonance imaging scan revealed polymicrogyria in frontal lobes and right Sylvian fissure.
Image 2: Short humerus with stippled epiphyses
Image 3: Short femur
Image 4: Mild right-sided ventriculomegaly
Image 5: Bilateral subependymal cysts
Video 1: Open and abnormal bullet shape of 4th ventricle, and shallow Sylvian fissure
Image 7: Polymicrogyria in frontal lobes
Image 8: Polymicrogyria in right Sylvian fissure
Image 9, Video 2, 3: Hepatosplenomegaly
The patient opted for clinical exome sequencing to screen for genetic causes. Clinical exome sequencing found homozygous point-nonsense mutation (NM_000318:c.373C>T (p.Arg123Ter)) in PEX2 gene. The peroxisomal biogenesis disorder Zellweger spectrum disorder with mutations in PEX2 gene was consistent with clinical diagnosis of this fetus. Counseling about the prognosis of Zellweger spectrum disorder was provided to the parents and termination was opted by both parents.
Discussion
Zellweger spectrum disorder (OMIM #214100), also known as cerebrohepatorenal syndrome, is an autosomal recessive disorder of peroxisome biogenesis and is defined by a continuum of phenotypes between “severe” and “mild.” Newborns with “severe” Zellweger spectrum disorder present with hypotonia, poor feeding, facial dysmorphism, neuronal migration defects associated with neonatal-onset seizures, renal cysts, bony stippling of the patella and long bones, and hepatomegaly. These neonates often die in the first year of life having made no developmental progress. Patients with the “intermediate / mild” form (previously called neonatal adrenoleukodystrophy and infantile Refsum disease) present with progressive sensory loss due to retinal dystrophy and sensorineural hearing loss, neurologic dysfunction with ataxia, leukodystrophy and polyneuropathy, as well as liver dysfunction, adrenal insufficiency, and renal oxalate stones. Progressive demyelination may lead to loss of previously acquired skills. Liver dysfunction may lead to vitamin K-responsive coagulopathy. Individuals with atypical Zellweger spectrum disorder do not present with sensory losses but have ataxia, peripheral neuropathy, and congenital cataracts [1].
The overall incidence of Zellweger spectrum disorder is about 1 in 133,000 births [1].
Zellweger spectrum disorder is caused by various mutations in PEX genes, which encode proteins called peroxins involved in peroxisome biogenesis. Peroxisomes are membrane-bound organelles involved in the α- and β-oxidation of fatty acids, the detoxification of glyoxylate and of reactive oxygen species, as well as the biosynthesis of ether phospholipids and bile acids, which are constituents of the cell membrane and myelin. Mutations in at least 13 different PEX genes have been associated with Zellweger spectrum disorder. The most common mutations are in the PEX1 or PEX6 gene, which is seen in approximately 60% and 15% of patients, respectively [2].
Zellweger spectrum disorder is characterized by increased accumulation of very long chain fatty acids in plasma and fibroblasts [3]. Elevated concentration of very long chain fatty acids is thought to cause adrenal cell dysfunction with decreased cortisol response and adrenal insufficiency [4]. Additionally, abnormal fatty acids, especially very long chain fatty acids, can be incorporated in neuronal cell membranes leading to injury and demyelination [5].
Prenatal diagnosis of Zellweger spectrum disorder is possible [6]. Ultrasound findings include subependymal pseudocysts [7], cerebral ventricular enlargement, renal hyperechogenicity [6, 8] and fetal hypokinesia [9]. MRI can be very helpful in suggesting the diagnosis by identifying migration anomalies such as focal pachygyria, periventricular leukodystrophy, and subependymal pseudocysts [6, 8, 10]. Definitive diagnosis can be made with whole exome sequencing.
The differential diagnosis for neuronal migration anomalies is infection with cytomegalovirus, as well as inborn errors of metabolism such as respiratory chain defect, and pyruvate dehydrogenase deficiency [8].
Zellweger spectrum disorder is a progressive disorder with a high mortality rate. With no curative treatment available, treatment options are limited to supportive care to improve quality of life. Because of its fatality in early life, prenatal diagnosis and genetic counseling for Zellweger spectrum disorder are crucial for planning care in future pregnancies [1].
References
[1] Steinberg SJ, Raymond GV, Braverman NE et al. “Zellweger Spectrum Disorder.” GeneReviews® [Internet]. Seattle: University of Washington; Publication date 12/2003 [Updated 10/2020]. https://www.ncbi.nlm.nih.gov/books/NBK1448/
[2] Waterham HR, Ebberink MS. Genetics and molecular basis of human peroxisome biogenesis disorders. Biochim Biophys Acta. 2012 Sep;1822(9):1430-41.
[3] Schutgens RB, Bouman IW, Nijenhuis AA, et al. Profiles of very-long-chain fatty acids in plasma, fibroblasts, and blood cells in Zellweger syndrome, X-linked adrenoleukodystrophy, and rhizomelic chondrodysplasia punctata. Clin Chem. 1993 Aug;39(8):1632-7.
[4] Berendse K, Engelen M, Linthorst GE, et al. High prevalence of primary adrenal insufficiency in Zellweger spectrum disorders. Orphanet J Rare Dis. 2014; 9: 133.
[5] Powers JM, Moser HW. Peroxisomal Disorders: Genotype, Phenotype, Major Neuropathologic Lesions, and Pathogenesis. Brain Pathol. 1998 Jan; 8(1): 101–120.
[6] Diaz J, Matsumoto L, Neville JK. “Prenatal diagnosis of zellweger syndrome by fetal MRI: a case report.” Radiol Case Rep. 2021 Oct 19;16(12):3950-3954
[7] Russel IM, van Sonderen L, van Straaten HL, et al. Subependymal germinolytic cysts in Zellweger syndrome. Pediatr Radiol. 1995;25(4):254-5.
[8] Mochel F, Grébille AG, Benachi A, et al. Contribution of fetal MR imaging in the prenatal diagnosis of Zellweger syndrome. AJNR Am J Neuroradiol. 2006 Feb;27(2):333-6.
[9] Johnson JM, Babul-Hirji R, Chitayat D. First-trimester increased nuchal translucency and fetal hypokinesia associated with Zellweger syndrome. Ultrasound Obstet Gynecol. 2001 Apr;17(4):344-6.
[10] Segers K, Pierquin G, Gaillez S, et al. Rapid prenatal diagnosis of fetal Zellweger syndrome by biochemical tests, complementation studies, and molecular analyses. Prenat Diagn. 2013 Feb;33(2):201-3.
Discussion Board
Winners

Fatih ULUC Turkey Physician

Umber Agarwal United Kingdom Maternal Fetal Medicine

Sara Abdallah Salem Egypt Physician

Halil Mesut Turkey Physician
Victoria Giang Viet Nam Physician