Case of the Week #531
Case of the Week #531
#1 -
09/16/2022
said
“I think that in this case, although meningocele is part of the present pathology, “cervical myelocystocele” can be considered the most complete and correct answer. I explain it below.
Regarding the images:
-Ultrasound (Images 1-6): axial, coronal and sagittal fetal sonograms demonstrate a cyst-within-a-cyst arising from the cervico-thoracic spine area, lying along the posterior aspect of the occiput. The posterior fossa is not properly visualized, thus the obliteration of the cisterna magna cannot be assessed.
-MRI (Images 7-8): sagittal T2-weighted image also demonstrating ‘cyst within a cyst’. Below the level of the defect, there is associated hydromyelia. Using this technique, a normal posterior fossa is identified, without signs suggestive of Chiari II.
Spinal dysraphisms result from derangement in the normal early embryonic development, between gestational weeks 2 and 6. The relevant steps in embryogenesis are gastrulation (weeks 2–3), primary neurulation (weeks 3–4), and secondary neurulation (weeks 5–6). The term dysraphism (from the Greek δυσ = bad and ραϕη = suture) etymologically refers to a defect of closure of the neural tube and, therefore, should apply to abnormalities of primary neurulation only. However, its use has been broadened to include all congenital spinal disorders in which there is anomalous differentiation and/or incomplete closure of dorsal midline structures: skin, muscles, vertebrae, meninges, and nervous tissue. Due to the common embryological origin, caudal spinal anomalies also are included in this group.
Spinal dysraphisms are classically categorized in two major subsets: open spinal dysraphisms (OSD) and closed spinal dysraphisms (CSD). OSDs are characterized by exposure of the nervous tissue and/or meninges to the environment through a congenital bony defect. Conversely, CSDs are covered by skin (there is no exposed neural tissue), although cutaneous stigmata, such as hairy nevus, capillary hemangioma, dimples, dyschromic patches, dystrophy, and subcutaneous masses, indicate their presence in as many as 50% of cases. CSDs are more numerous than OSDs, accounting for approximately two thirds of patients. The term “spina bifida” merely refers to defective fusion of posterior spinal bony elements but is widely used to refer to spinal dysraphisms in general. Four varieties of OSD exist (myelomeningocele, myelocele, hemimyelomeningocele, and hemimyelocele), with myelomeningocele encompassing 98.8% of cases. Usually, OSDs are located at the lumbar or lumbosacral level and are diagnosed antenatally by ultrasound. CSDs are much more heterogeneous than OSDs. In general, a large number of malformations belong to this group and some of them are not clinically evident at birth. In the vast majority of cases there is a subcutaneous mass at the lumbar or lumbosacral level, corresponding to a meningocele, lipomyelocele, lipomyelomeningocele, or terminal myelocystocele. At the cervico-thoracic level, CSDs with a subcutaneous mass are represented by the spectrum of nonterminal myelocystoceles.
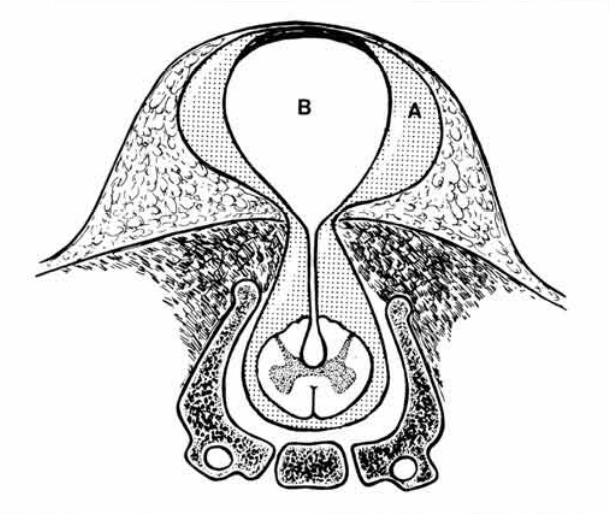
Cervical spinal dysraphism is rare, comprising only 3–8% of all cases of spinal dysraphism. It typically includes a cystic mass, which may be pedunculated or sessile, situated in the midline posteriorly and covered by skin, which is normal to thick at the base but thin and discoloured over the dome. There are two recognised subtypes: myelocystoceles and meningoceles. Steinbok and Cochrane hypothesize that the cervical meningocele and the myelocystocele are part of a spectrum of the same underlying developmental abnormality, namely limited dorsal myeloschisis, and the final presentation depends on the presence or absence of associated hydromyelia. The neural stalk, in continuity with the central canal, is considered the failure of the cutaneous ectoderm to separate from the neuroectoderm. If the central canal of the spinal canal is hydromyelic, the lumen of the neural stalk, which is in continuity with the central canal of the spinal cord, may be kept open, and fluid within the dorsal aspect of the stalk can distend the neuroglial tissue causing a dorsal sac. A meningocele sac surrounds the inner cyst and the space between these two sacs may be filled with cerebrospinal fluid in continuity with the subarachnoid space. The meningeal layer is covered by skin. In the absence of hydromyelia, the neural stalk narrows and regresses. The fibroneurovascular stalk may remain in a central position, as a band attaching the dorsal aspect of the spinal cord to the back of the meningocele sac, or may be displaced asymmetrically within the meningocele sac. This second form can be considered an “abortive” myelocystocele or “forme fruste” (Rossi type 1) versus previously described “complete” myelocystocele (Rossi type 2).
Myelocystocele is an occult spinal dysraphism in which the spinal cord and the arachnoid are herniated through a posterior spina bifida. It has been called syringocele and syringomyelocele. Sonography and MR imaging are useful in the evaluation of suspected myelocystoceles, both techniques showing the characteristic "cyst within a cyst" image. A thin stalk between the inner cyst and the spinal cord may be detected if the scan plane is perpendicular to the fetal spine and perpendicular to the stalk.
The Chiari II malformation is a complex congenital anomaly of the hindbrain characterized by a smaller than normal posterior cranial fossa with caudal displacement of the vermis, brainstem, and fourth ventricle. There is nearly universal association with open spinal dysraphisms with the exception of nonterminal myelocystoceles, which are the only form of closed spinal dysraphism that can be associated with a Chiari II malformation. Although all patients with open spinal dysraphisms present a Chiari II malformation, the reverse is not true. While >90% of patients with Chiari II malformation have open spinal defects, a minority of cases are discovered in patients with closed spinal dysraphisms (these are exceedingly rare myelocystoceles). It is important to examine the fetal head for signs of Chiari II malformation (obliterated cisterna magna, deformed cerebellum with 'banana sign', enlarged cerebral lateral ventricles, and concavity of the frontal bones with 'lemon sign').
Cervical spinal dysraphism is often accompanied by central nervous system anomalies other than Chiari II malformation, such as hydrocephalus, hydromyelia, lipomeningomyelocele, tethered cord and thickened filum, diastematomyelia, Klippel–Feil syndrome, thoracic hemivertebrae, and a dermal sinus or overlying cutaneous lesion. These children do not have obvious neurologic deficit at birth, but tend to have significant neurological deterioration and orthopedic problems with time, which is an argument to operate early. The purpose of surgical intervention is not only to remove the cystic mass for cosmetic reasons and to untether the cervical spinal cord prophylactically, but also to treat immediate neurological deficits.
Cervical myelocystocele, although rare, should be added to the traditional differential diagnosis when a cystic lesion of the posterior neck is demonstrated on prenatal ultrasound. A normal amniotic fluid alpha-fetoprotein and acetylcholinesterase levels reduces the likelihood of an open neural tube defect, but it is unlikely that a closed, simple meningocele with a small spinal defect would be distinguishable from a myelocystocele sonographically. The other common neck mass, cystic hygroma, is usually septate, extends laterally, and is often associated with fetal hydrops and chromosomal abnormalities.
Suggested readings:
1. Bhargava R, Hammond DI, Benzie RJ, Carlos E, Ventureyra G, Higgins MJ, Martin DJ. Prenatal demonstration of a cervical myelocystocele. Prenat Diagn. 1992 Aug;12(8):653-9. doi: 10.1002/pd.1970120806. PMID: 1279658.
2. Klein O, Coulomb MA, Ternier J, Lena G. Cervical myelocystocele: prenatal diagnosis and therapeutical considerations. Childs Nerv Syst. 2009 May;25(5):523-6. doi: 10.1007/s00381-008-0806-2. Epub 2009 Feb 11. PMID: 19212773.
3. Nishino A, Shirane R, So K, Arai H, Suzuki H, Sakurai Y. Cervical myelocystocele with Chiari II malformation: magnetic resonance imaging and surgical treatment. Surg Neurol. 1998 Mar;49(3):269-73. doi: 10.1016/s0090-3019(97)00180-8. PMID: 9508113.
4. Rossi A, Piatelli G, Gandolfo C, Pavanello M, Hoffmann C, Van Goethem JW, Cama A, Tortori-Donati P. Spectrum of nonterminal myelocystoceles. Neurosurgery. 2006 Mar;58(3):509-15; discussion 509-15. doi: 10.1227/01.NEU.0000197122.92954.82. PMID: 16528191.
5. Sauerbrei EE, Grant P. Prenatal diagnosis of myelocystoceles: report of two cases. J Ultrasound Med. 1999 Mar;18(3):247-52. doi: 10.7863/jum.1999.18.3.247. PMID: 10082361.
6. Steinbok P, Cochrane DD. Cervical meningoceles and myelocystoceles: a unifying hypothesis. Pediatr Neurosurg. 1995;23(6):317-22. doi: 10.1159/000120978. PMID: 8744001.
7. Talenti G, Giussani C, Parazzini C, Izzo G, Canonico F, Vergani P, Righini A. Early Prenatal MRI of Cervical "Abortive" Myelocystocele: Case Report and Review of the Literature. Neuropediatrics. 2017 Apr;48(2):104-107. doi: 10.1055/s-0036-1593985. Epub 2016 Nov 23. PMID: 27880967.
8. Tortori-Donati P, Rossi A, Biancheri R, and Cama A. Congenital Malformations of the Spine and Spinal Cord. In: Tortori-Donati P. Pediatric Neuroradiology: Brain, Head and Neck, Spine. Springer Berlin Heidelberg New York, 2005. Pag. 1551-1608
Images coming from reference 6: Steinbok P, Cochrane DD. Cervical meningoceles and myelocystoceles: a unifying hypothesis. Pediatr Neurosurg. 1995;23(6):317-22. doi: 10.1159/000120978. PMID: 8744001.
”
Regarding the images:
-Ultrasound (Images 1-6): axial, coronal and sagittal fetal sonograms demonstrate a cyst-within-a-cyst arising from the cervico-thoracic spine area, lying along the posterior aspect of the occiput. The posterior fossa is not properly visualized, thus the obliteration of the cisterna magna cannot be assessed.
-MRI (Images 7-8): sagittal T2-weighted image also demonstrating ‘cyst within a cyst’. Below the level of the defect, there is associated hydromyelia. Using this technique, a normal posterior fossa is identified, without signs suggestive of Chiari II.
Spinal dysraphisms result from derangement in the normal early embryonic development, between gestational weeks 2 and 6. The relevant steps in embryogenesis are gastrulation (weeks 2–3), primary neurulation (weeks 3–4), and secondary neurulation (weeks 5–6). The term dysraphism (from the Greek δυσ = bad and ραϕη = suture) etymologically refers to a defect of closure of the neural tube and, therefore, should apply to abnormalities of primary neurulation only. However, its use has been broadened to include all congenital spinal disorders in which there is anomalous differentiation and/or incomplete closure of dorsal midline structures: skin, muscles, vertebrae, meninges, and nervous tissue. Due to the common embryological origin, caudal spinal anomalies also are included in this group.
Spinal dysraphisms are classically categorized in two major subsets: open spinal dysraphisms (OSD) and closed spinal dysraphisms (CSD). OSDs are characterized by exposure of the nervous tissue and/or meninges to the environment through a congenital bony defect. Conversely, CSDs are covered by skin (there is no exposed neural tissue), although cutaneous stigmata, such as hairy nevus, capillary hemangioma, dimples, dyschromic patches, dystrophy, and subcutaneous masses, indicate their presence in as many as 50% of cases. CSDs are more numerous than OSDs, accounting for approximately two thirds of patients. The term “spina bifida” merely refers to defective fusion of posterior spinal bony elements but is widely used to refer to spinal dysraphisms in general. Four varieties of OSD exist (myelomeningocele, myelocele, hemimyelomeningocele, and hemimyelocele), with myelomeningocele encompassing 98.8% of cases. Usually, OSDs are located at the lumbar or lumbosacral level and are diagnosed antenatally by ultrasound. CSDs are much more heterogeneous than OSDs. In general, a large number of malformations belong to this group and some of them are not clinically evident at birth. In the vast majority of cases there is a subcutaneous mass at the lumbar or lumbosacral level, corresponding to a meningocele, lipomyelocele, lipomyelomeningocele, or terminal myelocystocele. At the cervico-thoracic level, CSDs with a subcutaneous mass are represented by the spectrum of nonterminal myelocystoceles.
Cervical spinal dysraphism is rare, comprising only 3–8% of all cases of spinal dysraphism. It typically includes a cystic mass, which may be pedunculated or sessile, situated in the midline posteriorly and covered by skin, which is normal to thick at the base but thin and discoloured over the dome. There are two recognised subtypes: myelocystoceles and meningoceles. Steinbok and Cochrane hypothesize that the cervical meningocele and the myelocystocele are part of a spectrum of the same underlying developmental abnormality, namely limited dorsal myeloschisis, and the final presentation depends on the presence or absence of associated hydromyelia. The neural stalk, in continuity with the central canal, is considered the failure of the cutaneous ectoderm to separate from the neuroectoderm. If the central canal of the spinal canal is hydromyelic, the lumen of the neural stalk, which is in continuity with the central canal of the spinal cord, may be kept open, and fluid within the dorsal aspect of the stalk can distend the neuroglial tissue causing a dorsal sac. A meningocele sac surrounds the inner cyst and the space between these two sacs may be filled with cerebrospinal fluid in continuity with the subarachnoid space. The meningeal layer is covered by skin. In the absence of hydromyelia, the neural stalk narrows and regresses. The fibroneurovascular stalk may remain in a central position, as a band attaching the dorsal aspect of the spinal cord to the back of the meningocele sac, or may be displaced asymmetrically within the meningocele sac. This second form can be considered an “abortive” myelocystocele or “forme fruste” (Rossi type 1) versus previously described “complete” myelocystocele (Rossi type 2).
Myelocystocele is an occult spinal dysraphism in which the spinal cord and the arachnoid are herniated through a posterior spina bifida. It has been called syringocele and syringomyelocele. Sonography and MR imaging are useful in the evaluation of suspected myelocystoceles, both techniques showing the characteristic "cyst within a cyst" image. A thin stalk between the inner cyst and the spinal cord may be detected if the scan plane is perpendicular to the fetal spine and perpendicular to the stalk.
The Chiari II malformation is a complex congenital anomaly of the hindbrain characterized by a smaller than normal posterior cranial fossa with caudal displacement of the vermis, brainstem, and fourth ventricle. There is nearly universal association with open spinal dysraphisms with the exception of nonterminal myelocystoceles, which are the only form of closed spinal dysraphism that can be associated with a Chiari II malformation. Although all patients with open spinal dysraphisms present a Chiari II malformation, the reverse is not true. While >90% of patients with Chiari II malformation have open spinal defects, a minority of cases are discovered in patients with closed spinal dysraphisms (these are exceedingly rare myelocystoceles). It is important to examine the fetal head for signs of Chiari II malformation (obliterated cisterna magna, deformed cerebellum with 'banana sign', enlarged cerebral lateral ventricles, and concavity of the frontal bones with 'lemon sign').
Cervical spinal dysraphism is often accompanied by central nervous system anomalies other than Chiari II malformation, such as hydrocephalus, hydromyelia, lipomeningomyelocele, tethered cord and thickened filum, diastematomyelia, Klippel–Feil syndrome, thoracic hemivertebrae, and a dermal sinus or overlying cutaneous lesion. These children do not have obvious neurologic deficit at birth, but tend to have significant neurological deterioration and orthopedic problems with time, which is an argument to operate early. The purpose of surgical intervention is not only to remove the cystic mass for cosmetic reasons and to untether the cervical spinal cord prophylactically, but also to treat immediate neurological deficits.
Cervical myelocystocele, although rare, should be added to the traditional differential diagnosis when a cystic lesion of the posterior neck is demonstrated on prenatal ultrasound. A normal amniotic fluid alpha-fetoprotein and acetylcholinesterase levels reduces the likelihood of an open neural tube defect, but it is unlikely that a closed, simple meningocele with a small spinal defect would be distinguishable from a myelocystocele sonographically. The other common neck mass, cystic hygroma, is usually septate, extends laterally, and is often associated with fetal hydrops and chromosomal abnormalities.
Suggested readings:
1. Bhargava R, Hammond DI, Benzie RJ, Carlos E, Ventureyra G, Higgins MJ, Martin DJ. Prenatal demonstration of a cervical myelocystocele. Prenat Diagn. 1992 Aug;12(8):653-9. doi: 10.1002/pd.1970120806. PMID: 1279658.
2. Klein O, Coulomb MA, Ternier J, Lena G. Cervical myelocystocele: prenatal diagnosis and therapeutical considerations. Childs Nerv Syst. 2009 May;25(5):523-6. doi: 10.1007/s00381-008-0806-2. Epub 2009 Feb 11. PMID: 19212773.
3. Nishino A, Shirane R, So K, Arai H, Suzuki H, Sakurai Y. Cervical myelocystocele with Chiari II malformation: magnetic resonance imaging and surgical treatment. Surg Neurol. 1998 Mar;49(3):269-73. doi: 10.1016/s0090-3019(97)00180-8. PMID: 9508113.
4. Rossi A, Piatelli G, Gandolfo C, Pavanello M, Hoffmann C, Van Goethem JW, Cama A, Tortori-Donati P. Spectrum of nonterminal myelocystoceles. Neurosurgery. 2006 Mar;58(3):509-15; discussion 509-15. doi: 10.1227/01.NEU.0000197122.92954.82. PMID: 16528191.
5. Sauerbrei EE, Grant P. Prenatal diagnosis of myelocystoceles: report of two cases. J Ultrasound Med. 1999 Mar;18(3):247-52. doi: 10.7863/jum.1999.18.3.247. PMID: 10082361.
6. Steinbok P, Cochrane DD. Cervical meningoceles and myelocystoceles: a unifying hypothesis. Pediatr Neurosurg. 1995;23(6):317-22. doi: 10.1159/000120978. PMID: 8744001.
7. Talenti G, Giussani C, Parazzini C, Izzo G, Canonico F, Vergani P, Righini A. Early Prenatal MRI of Cervical "Abortive" Myelocystocele: Case Report and Review of the Literature. Neuropediatrics. 2017 Apr;48(2):104-107. doi: 10.1055/s-0036-1593985. Epub 2016 Nov 23. PMID: 27880967.
8. Tortori-Donati P, Rossi A, Biancheri R, and Cama A. Congenital Malformations of the Spine and Spinal Cord. In: Tortori-Donati P. Pediatric Neuroradiology: Brain, Head and Neck, Spine. Springer Berlin Heidelberg New York, 2005. Pag. 1551-1608
Images coming from reference 6: Steinbok P, Cochrane DD. Cervical meningoceles and myelocystoceles: a unifying hypothesis. Pediatr Neurosurg. 1995;23(6):317-22. doi: 10.1159/000120978. PMID: 8744001.
”

#2 -
01/05/2023
I think that in this case, although meningocele is part of the present pathology, “cervical myelocystocele” can be considered the most complete and correct answer. I explain it below.
Regarding the images:
-Ultrasound (Images 1-6): axial, coronal and sagittal fetal sonograms demonstrate a cyst-within-a-cyst arising from the cervico-thoracic spine area, lying along the posterior aspect of the occiput. The posterior fossa is not properly visualized, thus the obliteration of the cisterna magna cannot be assessed.
-MRI (Images 7-8): sagittal T2-weighted image also demonstrating ‘cyst within a cyst’. Below the level of the defect, there is associated hydromyelia. Using this technique, a normal posterior fossa is identified, without signs suggestive of Chiari II.
Spinal dysraphisms result from derangement in the normal early embryonic development, between gestational weeks 2 and 6. The relevant steps in embryogenesis are gastrulation (weeks 2–3), primary neurulation (weeks 3–4), and secondary neurulation (weeks 5–6). The term dysraphism (from the Greek δυσ = bad and ραϕη = suture) etymologically refers to a defect of closure of the neural tube and, therefore, should apply to abnormalities of primary neurulation only. However, its use has been broadened to include all congenital spinal disorders in which there is anomalous differentiation and/or incomplete closure of dorsal midline structures: skin, muscles, vertebrae, meninges, and nervous tissue. Due to the common embryological origin, caudal spinal anomalies also are included in this group.
Spinal dysraphisms are classically categorized in two major subsets: open spinal dysraphisms (OSD) and closed spinal dysraphisms (CSD). OSDs are characterized by exposure of the nervous tissue and/or meninges to the environment through a congenital bony defect. Conversely, CSDs are covered by skin (there is no exposed neural tissue), although cutaneous stigmata, such as hairy nevus, capillary hemangioma, dimples, dyschromic patches, dystrophy, and subcutaneous masses, indicate their presence in as many as 50% of cases. CSDs are more numerous than OSDs, accounting for approximately two thirds of patients. The term “spina bifida” merely refers to defective fusion of posterior spinal bony elements but is widely used to refer to spinal dysraphisms in general. Four varieties of OSD exist (myelomeningocele, myelocele, hemimyelomeningocele, and hemimyelocele), with myelomeningocele encompassing 98.8% of cases. Usually, OSDs are located at the lumbar or lumbosacral level and are diagnosed antenatally by ultrasound. CSDs are much more heterogeneous than OSDs. In general, a large number of malformations belong to this group and some of them are not clinically evident at birth. In the vast majority of cases there is a subcutaneous mass at the lumbar or lumbosacral level, corresponding to a meningocele, lipomyelocele, lipomyelomeningocele, or terminal myelocystocele. At the cervico-thoracic level, CSDs with a subcutaneous mass are represented by the spectrum of nonterminal myelocystoceles.
Cervical spinal dysraphism is rare, comprising only 3–8% of all cases of spinal dysraphism. It typically includes a cystic mass, which may be pedunculated or sessile, situated in the midline posteriorly and covered by skin, which is normal to thick at the base but thin and discoloured over the dome. There are two recognised subtypes: myelocystoceles and meningoceles. Steinbok and Cochrane hypothesize that the cervical meningocele and the myelocystocele are part of a spectrum of the same underlying developmental abnormality, namely limited dorsal myeloschisis, and the final presentation depends on the presence or absence of associated hydromyelia. The neural stalk, in continuity with the central canal, is considered the failure of the cutaneous ectoderm to separate from the neuroectoderm. If the central canal of the spinal canal is hydromyelic, the lumen of the neural stalk, which is in continuity with the central canal of the spinal cord, may be kept open, and fluid within the dorsal aspect of the stalk can distend the neuroglial tissue causing a dorsal sac. A meningocele sac surrounds the inner cyst and the space between these two sacs may be filled with cerebrospinal fluid in continuity with the subarachnoid space. The meningeal layer is covered by skin. In the absence of hydromyelia, the neural stalk narrows and regresses. The fibroneurovascular stalk may remain in a central position, as a band attaching the dorsal aspect of the spinal cord to the back of the meningocele sac, or may be displaced asymmetrically within the meningocele sac. This second form can be considered an “abortive” myelocystocele or “forme fruste” (Rossi type 1) versus previously described “complete” myelocystocele (Rossi type 2).
Myelocystocele is an occult spinal dysraphism in which the spinal cord and the arachnoid are herniated through a posterior spina bifida. It has been called syringocele and syringomyelocele. Sonography and MR imaging are useful in the evaluation of suspected myelocystoceles, both techniques showing the characteristic "cyst within a cyst" image. A thin stalk between the inner cyst and the spinal cord may be detected if the scan plane is perpendicular to the fetal spine and perpendicular to the stalk.
The Chiari II malformation is a complex congenital anomaly of the hindbrain characterized by a smaller than normal posterior cranial fossa with caudal displacement of the vermis, brainstem, and fourth ventricle. There is nearly universal association with open spinal dysraphisms with the exception of nonterminal myelocystoceles, which are the only form of closed spinal dysraphism that can be associated with a Chiari II malformation. Although all patients with open spinal dysraphisms present a Chiari II malformation, the reverse is not true. While >90% of patients with Chiari II malformation have open spinal defects, a minority of cases are discovered in patients with closed spinal dysraphisms (these are exceedingly rare myelocystoceles). It is important to examine the fetal head for signs of Chiari II malformation (obliterated cisterna magna, deformed cerebellum with 'banana sign', enlarged cerebral lateral ventricles, and concavity of the frontal bones with 'lemon sign').
Cervical spinal dysraphism is often accompanied by central nervous system anomalies other than Chiari II malformation, such as hydrocephalus, hydromyelia, lipomeningomyelocele, tethered cord and thickened filum, diastematomyelia, Klippel–Feil syndrome, thoracic hemivertebrae, and a dermal sinus or overlying cutaneous lesion. These children do not have obvious neurologic deficit at birth, but tend to have significant neurological deterioration and orthopedic problems with time, which is an argument to operate early. The purpose of surgical intervention is not only to remove the cystic mass for cosmetic reasons and to untether the cervical spinal cord prophylactically, but also to treat immediate neurological deficits.
Cervical myelocystocele, although rare, should be added to the traditional differential diagnosis when a cystic lesion of the posterior neck is demonstrated on prenatal ultrasound. A normal amniotic fluid alpha-fetoprotein and acetylcholinesterase levels reduces the likelihood of an open neural tube defect, but it is unlikely that a closed, simple meningocele with a small spinal defect would be distinguishable from a myelocystocele sonographically. The other common neck mass, cystic hygroma, is usually septate, extends laterally, and is often associated with fetal hydrops and chromosomal abnormalities.
Suggested readings:
1. Bhargava R, Hammond DI, Benzie RJ, Carlos E, Ventureyra G, Higgins MJ, Martin DJ. Prenatal demonstration of a cervical myelocystocele. Prenat Diagn. 1992 Aug;12(8):653-9. doi: 10.1002/pd.1970120806. PMID: 1279658.
2. Klein O, Coulomb MA, Ternier J, Lena G. Cervical myelocystocele: prenatal diagnosis and therapeutical considerations. Childs Nerv Syst. 2009 May;25(5):523-6. doi: 10.1007/s00381-008-0806-2. Epub 2009 Feb 11. PMID: 19212773.
3. Nishino A, Shirane R, So K, Arai H, Suzuki H, Sakurai Y. Cervical myelocystocele with Chiari II malformation: magnetic resonance imaging and surgical treatment. Surg Neurol. 1998 Mar;49(3):269-73. doi: 10.1016/s0090-3019(97)00180-8. PMID: 9508113.
4. Rossi A, Piatelli G, Gandolfo C, Pavanello M, Hoffmann C, Van Goethem JW, Cama A, Tortori-Donati P. Spectrum of nonterminal myelocystoceles. Neurosurgery. 2006 Mar;58(3):509-15; discussion 509-15. doi: 10.1227/01.NEU.0000197122.92954.82. PMID: 16528191.
5. Sauerbrei EE, Grant P. Prenatal diagnosis of myelocystoceles: report of two cases. J Ultrasound Med. 1999 Mar;18(3):247-52. doi: 10.7863/jum.1999.18.3.247. PMID: 10082361.
6. Steinbok P, Cochrane DD. Cervical meningoceles and myelocystoceles: a unifying hypothesis. Pediatr Neurosurg. 1995;23(6):317-22. doi: 10.1159/000120978. PMID: 8744001.
7. Talenti G, Giussani C, Parazzini C, Izzo G, Canonico F, Vergani P, Righini A. Early Prenatal MRI of Cervical "Abortive" Myelocystocele: Case Report and Review of the Literature. Neuropediatrics. 2017 Apr;48(2):104-107. doi: 10.1055/s-0036-1593985. Epub 2016 Nov 23. PMID: 27880967.
8. Tortori-Donati P, Rossi A, Biancheri R, and Cama A. Congenital Malformations of the Spine and Spinal Cord. In: Tortori-Donati P. Pediatric Neuroradiology: Brain, Head and Neck, Spine. Springer Berlin Heidelberg New York, 2005. Pag. 1551-1608
Images coming from reference 6: Steinbok P, Cochrane DD. Cervical meningoceles and myelocystoceles: a unifying hypothesis. Pediatr Neurosurg. 1995;23(6):317-22. doi: 10.1159/000120978. PMID: 8744001.
said
“What a great text Sir. Clear and easy to understand. Thank You”
#3 -
01/15/2023
I think that in this case, although meningocele is part of the present pathology, “cervical myelocystocele” can be considered the most complete and correct answer. I explain it below.
Regarding the images:
-Ultrasound (Images 1-6): axial, coronal and sagittal fetal sonograms demonstrate a cyst-within-a-cyst arising from the cervico-thoracic spine area, lying along the posterior aspect of the occiput. The posterior fossa is not properly visualized, thus the obliteration of the cisterna magna cannot be assessed.
-MRI (Images 7-8): sagittal T2-weighted image also demonstrating ‘cyst within a cyst’. Below the level of the defect, there is associated hydromyelia. Using this technique, a normal posterior fossa is identified, without signs suggestive of Chiari II.
Spinal dysraphisms result from derangement in the normal early embryonic development, between gestational weeks 2 and 6. The relevant steps in embryogenesis are gastrulation (weeks 2–3), primary neurulation (weeks 3–4), and secondary neurulation (weeks 5–6). The term dysraphism (from the Greek δυσ = bad and ραϕη = suture) etymologically refers to a defect of closure of the neural tube and, therefore, should apply to abnormalities of primary neurulation only. However, its use has been broadened to include all congenital spinal disorders in which there is anomalous differentiation and/or incomplete closure of dorsal midline structures: skin, muscles, vertebrae, meninges, and nervous tissue. Due to the common embryological origin, caudal spinal anomalies also are included in this group.
Spinal dysraphisms are classically categorized in two major subsets: open spinal dysraphisms (OSD) and closed spinal dysraphisms (CSD). OSDs are characterized by exposure of the nervous tissue and/or meninges to the environment through a congenital bony defect. Conversely, CSDs are covered by skin (there is no exposed neural tissue), although cutaneous stigmata, such as hairy nevus, capillary hemangioma, dimples, dyschromic patches, dystrophy, and subcutaneous masses, indicate their presence in as many as 50% of cases. CSDs are more numerous than OSDs, accounting for approximately two thirds of patients. The term “spina bifida” merely refers to defective fusion of posterior spinal bony elements but is widely used to refer to spinal dysraphisms in general. Four varieties of OSD exist (myelomeningocele, myelocele, hemimyelomeningocele, and hemimyelocele), with myelomeningocele encompassing 98.8% of cases. Usually, OSDs are located at the lumbar or lumbosacral level and are diagnosed antenatally by ultrasound. CSDs are much more heterogeneous than OSDs. In general, a large number of malformations belong to this group and some of them are not clinically evident at birth. In the vast majority of cases there is a subcutaneous mass at the lumbar or lumbosacral level, corresponding to a meningocele, lipomyelocele, lipomyelomeningocele, or terminal myelocystocele. At the cervico-thoracic level, CSDs with a subcutaneous mass are represented by the spectrum of nonterminal myelocystoceles.
Cervical spinal dysraphism is rare, comprising only 3–8% of all cases of spinal dysraphism. It typically includes a cystic mass, which may be pedunculated or sessile, situated in the midline posteriorly and covered by skin, which is normal to thick at the base but thin and discoloured over the dome. There are two recognised subtypes: myelocystoceles and meningoceles. Steinbok and Cochrane hypothesize that the cervical meningocele and the myelocystocele are part of a spectrum of the same underlying developmental abnormality, namely limited dorsal myeloschisis, and the final presentation depends on the presence or absence of associated hydromyelia. The neural stalk, in continuity with the central canal, is considered the failure of the cutaneous ectoderm to separate from the neuroectoderm. If the central canal of the spinal canal is hydromyelic, the lumen of the neural stalk, which is in continuity with the central canal of the spinal cord, may be kept open, and fluid within the dorsal aspect of the stalk can distend the neuroglial tissue causing a dorsal sac. A meningocele sac surrounds the inner cyst and the space between these two sacs may be filled with cerebrospinal fluid in continuity with the subarachnoid space. The meningeal layer is covered by skin. In the absence of hydromyelia, the neural stalk narrows and regresses. The fibroneurovascular stalk may remain in a central position, as a band attaching the dorsal aspect of the spinal cord to the back of the meningocele sac, or may be displaced asymmetrically within the meningocele sac. This second form can be considered an “abortive” myelocystocele or “forme fruste” (Rossi type 1) versus previously described “complete” myelocystocele (Rossi type 2).
Myelocystocele is an occult spinal dysraphism in which the spinal cord and the arachnoid are herniated through a posterior spina bifida. It has been called syringocele and syringomyelocele. Sonography and MR imaging are useful in the evaluation of suspected myelocystoceles, both techniques showing the characteristic "cyst within a cyst" image. A thin stalk between the inner cyst and the spinal cord may be detected if the scan plane is perpendicular to the fetal spine and perpendicular to the stalk.
The Chiari II malformation is a complex congenital anomaly of the hindbrain characterized by a smaller than normal posterior cranial fossa with caudal displacement of the vermis, brainstem, and fourth ventricle. There is nearly universal association with open spinal dysraphisms with the exception of nonterminal myelocystoceles, which are the only form of closed spinal dysraphism that can be associated with a Chiari II malformation. Although all patients with open spinal dysraphisms present a Chiari II malformation, the reverse is not true. While >90% of patients with Chiari II malformation have open spinal defects, a minority of cases are discovered in patients with closed spinal dysraphisms (these are exceedingly rare myelocystoceles). It is important to examine the fetal head for signs of Chiari II malformation (obliterated cisterna magna, deformed cerebellum with 'banana sign', enlarged cerebral lateral ventricles, and concavity of the frontal bones with 'lemon sign').
Cervical spinal dysraphism is often accompanied by central nervous system anomalies other than Chiari II malformation, such as hydrocephalus, hydromyelia, lipomeningomyelocele, tethered cord and thickened filum, diastematomyelia, Klippel–Feil syndrome, thoracic hemivertebrae, and a dermal sinus or overlying cutaneous lesion. These children do not have obvious neurologic deficit at birth, but tend to have significant neurological deterioration and orthopedic problems with time, which is an argument to operate early. The purpose of surgical intervention is not only to remove the cystic mass for cosmetic reasons and to untether the cervical spinal cord prophylactically, but also to treat immediate neurological deficits.
Cervical myelocystocele, although rare, should be added to the traditional differential diagnosis when a cystic lesion of the posterior neck is demonstrated on prenatal ultrasound. A normal amniotic fluid alpha-fetoprotein and acetylcholinesterase levels reduces the likelihood of an open neural tube defect, but it is unlikely that a closed, simple meningocele with a small spinal defect would be distinguishable from a myelocystocele sonographically. The other common neck mass, cystic hygroma, is usually septate, extends laterally, and is often associated with fetal hydrops and chromosomal abnormalities.
Suggested readings:
1. Bhargava R, Hammond DI, Benzie RJ, Carlos E, Ventureyra G, Higgins MJ, Martin DJ. Prenatal demonstration of a cervical myelocystocele. Prenat Diagn. 1992 Aug;12(8):653-9. doi: 10.1002/pd.1970120806. PMID: 1279658.
2. Klein O, Coulomb MA, Ternier J, Lena G. Cervical myelocystocele: prenatal diagnosis and therapeutical considerations. Childs Nerv Syst. 2009 May;25(5):523-6. doi: 10.1007/s00381-008-0806-2. Epub 2009 Feb 11. PMID: 19212773.
3. Nishino A, Shirane R, So K, Arai H, Suzuki H, Sakurai Y. Cervical myelocystocele with Chiari II malformation: magnetic resonance imaging and surgical treatment. Surg Neurol. 1998 Mar;49(3):269-73. doi: 10.1016/s0090-3019(97)00180-8. PMID: 9508113.
4. Rossi A, Piatelli G, Gandolfo C, Pavanello M, Hoffmann C, Van Goethem JW, Cama A, Tortori-Donati P. Spectrum of nonterminal myelocystoceles. Neurosurgery. 2006 Mar;58(3):509-15; discussion 509-15. doi: 10.1227/01.NEU.0000197122.92954.82. PMID: 16528191.
5. Sauerbrei EE, Grant P. Prenatal diagnosis of myelocystoceles: report of two cases. J Ultrasound Med. 1999 Mar;18(3):247-52. doi: 10.7863/jum.1999.18.3.247. PMID: 10082361.
6. Steinbok P, Cochrane DD. Cervical meningoceles and myelocystoceles: a unifying hypothesis. Pediatr Neurosurg. 1995;23(6):317-22. doi: 10.1159/000120978. PMID: 8744001.
7. Talenti G, Giussani C, Parazzini C, Izzo G, Canonico F, Vergani P, Righini A. Early Prenatal MRI of Cervical "Abortive" Myelocystocele: Case Report and Review of the Literature. Neuropediatrics. 2017 Apr;48(2):104-107. doi: 10.1055/s-0036-1593985. Epub 2016 Nov 23. PMID: 27880967.
8. Tortori-Donati P, Rossi A, Biancheri R, and Cama A. Congenital Malformations of the Spine and Spinal Cord. In: Tortori-Donati P. Pediatric Neuroradiology: Brain, Head and Neck, Spine. Springer Berlin Heidelberg New York, 2005. Pag. 1551-1608
Images coming from reference 6: Steinbok P, Cochrane DD. Cervical meningoceles and myelocystoceles: a unifying hypothesis. Pediatr Neurosurg. 1995;23(6):317-22. doi: 10.1159/000120978. PMID: 8744001.
said
“Thank you very much for your comment. I'm glad you liked it.”
#4 -
09/10/2023
I think that in this case, although meningocele is part of the present pathology, “cervical myelocystocele” can be considered the most complete and correct answer. I explain it below.
Regarding the images:
-Ultrasound (Images 1-6): axial, coronal and sagittal fetal sonograms demonstrate a cyst-within-a-cyst arising from the cervico-thoracic spine area, lying along the posterior aspect of the occiput. The posterior fossa is not properly visualized, thus the obliteration of the cisterna magna cannot be assessed.
-MRI (Images 7-8): sagittal T2-weighted image also demonstrating ‘cyst within a cyst’. Below the level of the defect, there is associated hydromyelia. Using this technique, a normal posterior fossa is identified, without signs suggestive of Chiari II.
Spinal dysraphisms result from derangement in the normal early embryonic development, between gestational weeks 2 and 6. The relevant steps in embryogenesis are gastrulation (weeks 2–3), primary neurulation (weeks 3–4), and secondary neurulation (weeks 5–6). The term dysraphism (from the Greek δυσ = bad and ραϕη = suture) etymologically refers to a defect of closure of the neural tube and, therefore, should apply to abnormalities of primary neurulation only. However, its use has been broadened to include all congenital spinal disorders in which there is anomalous differentiation and/or incomplete closure of dorsal midline structures: skin, muscles, vertebrae, meninges, and nervous tissue. Due to the common embryological origin, caudal spinal anomalies also are included in this group.
Spinal dysraphisms are classically categorized in two major subsets: open spinal dysraphisms (OSD) and closed spinal dysraphisms (CSD). OSDs are characterized by exposure of the nervous tissue and/or meninges to the environment through a congenital bony defect. Conversely, CSDs are covered by skin (there is no exposed neural tissue), although cutaneous stigmata, such as hairy nevus, capillary hemangioma, dimples, dyschromic patches, dystrophy, and subcutaneous masses, indicate their presence in as many as 50% of cases. CSDs are more numerous than OSDs, accounting for approximately two thirds of patients. The term “spina bifida” merely refers to defective fusion of posterior spinal bony elements but is widely used to refer to spinal dysraphisms in general. Four varieties of OSD exist (myelomeningocele, myelocele, hemimyelomeningocele, and hemimyelocele), with myelomeningocele encompassing 98.8% of cases. Usually, OSDs are located at the lumbar or lumbosacral level and are diagnosed antenatally by ultrasound. CSDs are much more heterogeneous than OSDs. In general, a large number of malformations belong to this group and some of them are not clinically evident at birth. In the vast majority of cases there is a subcutaneous mass at the lumbar or lumbosacral level, corresponding to a meningocele, lipomyelocele, lipomyelomeningocele, or terminal myelocystocele. At the cervico-thoracic level, CSDs with a subcutaneous mass are represented by the spectrum of nonterminal myelocystoceles.
Cervical spinal dysraphism is rare, comprising only 3–8% of all cases of spinal dysraphism. It typically includes a cystic mass, which may be pedunculated or sessile, situated in the midline posteriorly and covered by skin, which is normal to thick at the base but thin and discoloured over the dome. There are two recognised subtypes: myelocystoceles and meningoceles. Steinbok and Cochrane hypothesize that the cervical meningocele and the myelocystocele are part of a spectrum of the same underlying developmental abnormality, namely limited dorsal myeloschisis, and the final presentation depends on the presence or absence of associated hydromyelia. The neural stalk, in continuity with the central canal, is considered the failure of the cutaneous ectoderm to separate from the neuroectoderm. If the central canal of the spinal canal is hydromyelic, the lumen of the neural stalk, which is in continuity with the central canal of the spinal cord, may be kept open, and fluid within the dorsal aspect of the stalk can distend the neuroglial tissue causing a dorsal sac. A meningocele sac surrounds the inner cyst and the space between these two sacs may be filled with cerebrospinal fluid in continuity with the subarachnoid space. The meningeal layer is covered by skin. In the absence of hydromyelia, the neural stalk narrows and regresses. The fibroneurovascular stalk may remain in a central position, as a band attaching the dorsal aspect of the spinal cord to the back of the meningocele sac, or may be displaced asymmetrically within the meningocele sac. This second form can be considered an “abortive” myelocystocele or “forme fruste” (Rossi type 1) versus previously described “complete” myelocystocele (Rossi type 2).
Myelocystocele is an occult spinal dysraphism in which the spinal cord and the arachnoid are herniated through a posterior spina bifida. It has been called syringocele and syringomyelocele. Sonography and MR imaging are useful in the evaluation of suspected myelocystoceles, both techniques showing the characteristic "cyst within a cyst" image. A thin stalk between the inner cyst and the spinal cord may be detected if the scan plane is perpendicular to the fetal spine and perpendicular to the stalk.
The Chiari II malformation is a complex congenital anomaly of the hindbrain characterized by a smaller than normal posterior cranial fossa with caudal displacement of the vermis, brainstem, and fourth ventricle. There is nearly universal association with open spinal dysraphisms with the exception of nonterminal myelocystoceles, which are the only form of closed spinal dysraphism that can be associated with a Chiari II malformation. Although all patients with open spinal dysraphisms present a Chiari II malformation, the reverse is not true. While >90% of patients with Chiari II malformation have open spinal defects, a minority of cases are discovered in patients with closed spinal dysraphisms (these are exceedingly rare myelocystoceles). It is important to examine the fetal head for signs of Chiari II malformation (obliterated cisterna magna, deformed cerebellum with 'banana sign', enlarged cerebral lateral ventricles, and concavity of the frontal bones with 'lemon sign').
Cervical spinal dysraphism is often accompanied by central nervous system anomalies other than Chiari II malformation, such as hydrocephalus, hydromyelia, lipomeningomyelocele, tethered cord and thickened filum, diastematomyelia, Klippel–Feil syndrome, thoracic hemivertebrae, and a dermal sinus or overlying cutaneous lesion. These children do not have obvious neurologic deficit at birth, but tend to have significant neurological deterioration and orthopedic problems with time, which is an argument to operate early. The purpose of surgical intervention is not only to remove the cystic mass for cosmetic reasons and to untether the cervical spinal cord prophylactically, but also to treat immediate neurological deficits.
Cervical myelocystocele, although rare, should be added to the traditional differential diagnosis when a cystic lesion of the posterior neck is demonstrated on prenatal ultrasound. A normal amniotic fluid alpha-fetoprotein and acetylcholinesterase levels reduces the likelihood of an open neural tube defect, but it is unlikely that a closed, simple meningocele with a small spinal defect would be distinguishable from a myelocystocele sonographically. The other common neck mass, cystic hygroma, is usually septate, extends laterally, and is often associated with fetal hydrops and chromosomal abnormalities.
Suggested readings:
1. Bhargava R, Hammond DI, Benzie RJ, Carlos E, Ventureyra G, Higgins MJ, Martin DJ. Prenatal demonstration of a cervical myelocystocele. Prenat Diagn. 1992 Aug;12(8):653-9. doi: 10.1002/pd.1970120806. PMID: 1279658.
2. Klein O, Coulomb MA, Ternier J, Lena G. Cervical myelocystocele: prenatal diagnosis and therapeutical considerations. Childs Nerv Syst. 2009 May;25(5):523-6. doi: 10.1007/s00381-008-0806-2. Epub 2009 Feb 11. PMID: 19212773.
3. Nishino A, Shirane R, So K, Arai H, Suzuki H, Sakurai Y. Cervical myelocystocele with Chiari II malformation: magnetic resonance imaging and surgical treatment. Surg Neurol. 1998 Mar;49(3):269-73. doi: 10.1016/s0090-3019(97)00180-8. PMID: 9508113.
4. Rossi A, Piatelli G, Gandolfo C, Pavanello M, Hoffmann C, Van Goethem JW, Cama A, Tortori-Donati P. Spectrum of nonterminal myelocystoceles. Neurosurgery. 2006 Mar;58(3):509-15; discussion 509-15. doi: 10.1227/01.NEU.0000197122.92954.82. PMID: 16528191.
5. Sauerbrei EE, Grant P. Prenatal diagnosis of myelocystoceles: report of two cases. J Ultrasound Med. 1999 Mar;18(3):247-52. doi: 10.7863/jum.1999.18.3.247. PMID: 10082361.
6. Steinbok P, Cochrane DD. Cervical meningoceles and myelocystoceles: a unifying hypothesis. Pediatr Neurosurg. 1995;23(6):317-22. doi: 10.1159/000120978. PMID: 8744001.
7. Talenti G, Giussani C, Parazzini C, Izzo G, Canonico F, Vergani P, Righini A. Early Prenatal MRI of Cervical "Abortive" Myelocystocele: Case Report and Review of the Literature. Neuropediatrics. 2017 Apr;48(2):104-107. doi: 10.1055/s-0036-1593985. Epub 2016 Nov 23. PMID: 27880967.
8. Tortori-Donati P, Rossi A, Biancheri R, and Cama A. Congenital Malformations of the Spine and Spinal Cord. In: Tortori-Donati P. Pediatric Neuroradiology: Brain, Head and Neck, Spine. Springer Berlin Heidelberg New York, 2005. Pag. 1551-1608
Images coming from reference 6: Steinbok P, Cochrane DD. Cervical meningoceles and myelocystoceles: a unifying hypothesis. Pediatr Neurosurg. 1995;23(6):317-22. doi: 10.1159/000120978. PMID: 8744001.
said
“The reel diagnosis was: a LDM: Limited Dorsal Myeloschisis; operated on, the child is progressing well until the latest news
”
”
Bronshtein Moshe